
Abstract
Four different cDNA libraries were constructed from sunflower protoplasts growing under embryogenic and non-embryogenic conditions: one standard library from each condition and two subtractive libraries in opposite sense. A total of 22,876 cDNA clones were obtained and 4800 ESTs were sequenced, giving rise to 2479 high quality ESTs representing an unigene set of 1502 sequences. This set was compared with ESTs represented in public databases using the programs BLASTN and BLASTX, and its members were classified according to putative function using the catalog in the Kyoto Encyclopedia of Genes and Genomes (KEGG). Some 33% of sequences failed to align with existing plant ESTs and therefore represent putative novel genes. The libraries show a low level of redundancy and, on average, 50% of the present ESTs have not been previously reported for sunflower. Several potentially interesting genes were identified, based on their homology with genes involved in animal zygotic division or plant embryogenesis. We also identified two ESTs that show significantly different levels of expression under embryogenic and non-embryogenic conditions. The libraries described here represent an original and valuable resource for the discovery of yet unknown genes putatively involved in dicot embryogenesis and improving our knowledge of the mechanisms involved in polarity acquisition by plant embryos.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The structural establishment of the dicotyledonous (dicot) embryo occurs under the influence of genes that control complex networks of molecular interactions. Although several genes implicated in embryo development have been identified in recent years (Jürgens et al. 1997; Souter and Lindsey 2000; Chaudhury et al. 2001; Jurgens 2001), the puzzle is still far from being fully assembled. Arabidopsis thaliana mutants that display abnormal embryo development have greatly facilitated the identification of genes expressed in the developing embryo (Torres-Ruiz and Jürgens 1994; Busch et al. 1996; Hardtke and Berleth 1998; Chen et al. 2001). However, although the morphological course of pattern development in Arabidopsis is well known (West and Harada 1993; Goldberg et al. 1994; Jürgens 1995, 2001), analysis of the early stages of embryogenesis at the molecular level is difficult, because the zygote is tightly surrounded by maternal tissues. In angiosperms, embryogenesis begins with a double fertilization event: pollen nuclei fertilize the egg cell and the central cell to form the zygote and the endosperm, respectively. The zygote then divides asymmetrically to give rise to a large basal cell and a small apical cell. The basal cell forms the suspensor, an embryonic structure that senesces during early embryogenesis, leaving only the uppermost cell, which becomes part of the embryonic root. The apical cell develops into the embryo proper. Embryo development proceeds along two main axes: apical-basal and radial. Since the young embryo develops deep within the maternal tissues, making it very difficult to investigate using conventional methods (West and Harada 1993; Goldberg et al. 1994; Jürgens 2001), alternative approaches, such as the study of somatic embryogenesis or the use of Fucus (an alga) as a model (Zimmerman 1993; Belanger and Quatrano 2000), have sometimes been adopted.
In recent years, Helianthus annuus , a member of the Compositae originating from North America but domesticated and easily bred in Europe, has been proposed as a putative supplementary model to A. thaliana for the study of dicot embryogenesis, owing to the synchronized, flowering of its inflorescences and the large size of its embryos. Several tools have been developed recently, making the study of this agriculturally interesting plant even easier. Genetic linkage maps are now available for almost the entire sunflower genome (Flores Berrios et al. 2000; Tang et al. 2002). In addition, two BAC libraries have been described (Caboche and Boucly 2000; Gentzbittel et al. 2002). Concomitantly, large-scale EST sequencing programs have been carried out, giving rise to 47,000 entries in Genbank. A collection of 2000 irradiated mutants is being screened for plants showing abnormal embryo development, by adopting the phenotypic screen described by Meinke (1985). Dicot embryogenesis is remarkably conserved in exalbuminous plants (plants without endosperm in the seed) and only minor differences exist between the development of A. thaliana and sunflower embryos. Moreover, in vitro somatic embryogenesis in sunflower could be used as a model for studying the mechanism of the first asymmetric zygotic division (Alibert et al. 1994). In the sunflower system, an isolated cell (a protoplast) can either develop into somatic tissue or give rise to an embryoid structure depending on culture conditions. When protoplasts are cultured in liquid medium they divide symmetrically and form a callus (the non-embryogenic condition); in contrast, when they are cultured embedded in agarose they divide asymmetrically and evolve into an embryoid structure (the embryogenic condition) (Chanabe et al. 1989). This asymmetrical division has been proposed as a functional model for the first asymmetrical division in the fertilized zygote. This model also has several advantages, such as the absence of maternal tissue, synchronization of the cell divisions and the fact that it is much easier to collect large amounts of embryogenic material than by embryos (which must be obtained by dissection).
This study is the first medium-scale sequencing project aimed at gaining information on gene expression profiles during the development of the sunflower embryo. Specifically, we focused on the first asymmetric division of the zygote, which appears to be crucial for the correct establishment of pattern development in the embryo. For example, gnom mutants of A. thaliana, which are defective in establishing the asymmetry of the first zygotic division, subsequently show aberrant apical-basal patterning and a very severe mutant phenotype in the adult plant (Mayer et al. 1993). The strategy of cDNA library subtraction was chosen because it has already been successful in identifying genes that are preferentially or specifically expressed in small amounts of cells or tissues (Robertson et al. 1994). We thus created two Suppressive Subtractive Hybridization (SSH) libraries enriched, respectively, in genes expressed under embryogenic or non-embryogenic conditions. We also created standard reference libraries from protoplasts cultured in embryogenic and non-embryogenic conditions, respectively. The classification of the ESTs into functional groups is expected to give a general idea of the different functions that are required under both types of condition, and differential representation of transcripts in the two reference libraries can be used to infer what genes are differentially expressed. The main aims of this study were to obtain an initial overview of the genes expressed under embryogenic conditions, and to extend our knowledge of asymmetrical division mechanisms by combining the analysis of reference and subtractive libraries. We describe a novel set of cDNA resources that are of interest for developmental genomics studies not only in sunflower but also in other exalbuminous dicots.
Materials and methods
Plant materials
The sunflower Emil hybrid (Pioneer Hi bred Inc.) was used in this study. Hypocotyl protoplasts were isolated and cultured according to Chanabé et al. (1989, 1991). Protoplasts were cultured at a final density of 106/ml in either liquid medium or 0.5% low-melting-point agarose (Sea Plaque; FMC Bioproducts). Protoplasts were divided into five identical portions. One portion was sampled on each day of culture.
cDNA library construction
RNA was isolated according to Chomczynski and Sacchi 1987. For construction of the reference library from protoplasts cultured under the non-embryogenic condition (HaDplR2) mRNAs were isolated using the Messagemaker Reagent Assembly (Invitrogen) according to the manufacturer’s instructions. The cDNA library was generated using the Superscript Plasmid System for cDNA Synthesis and Plasmid Cloning (Invitrogen) according to the manufacturer’s instructions. This library was cloned in pSPORT2.
The reference cDNA library from protoplasts kept under embryogenic conditions (HaDpsR1) was generated from total RNA using a Smart cDNA Library Construction kit (Clontech) according to the manufacturer’s instructions. cDNAs were cloned in the pTriplEx2 vector by in vivo excision. Reciprocal subtraction libraries (HaSemS3, HaSemS4) were created using the PCR-Select cDNA Subtraction kit (Clontech) according to the manufacturer’s conditions. cDNAs were cloned in pCRII using the TA Cloning kit (Invitrogen) according to the manufacturer’s instructions.
In each case except one, ligated cDNA was introduced into Escherichia coli UltraMAX DH5 α-FT competent cells by electroporation using a Gene Pulser II (Bio-Rad); the exception was HaDpsR1, which was cloned in E. coli BM25-8.
Expressed Sequence Tag (EST) sequencing and data analysis
Sequencing experiments were carried on 1632 cDNA clones from HaDplR2, 1056 clones from HaSemS3, 1056 clones from HaSemS4 and 1056 clones from the HaDpsR1 library. Library sequencing was performed using BigDye Terminator v2.0 and v3.0 (Applied Biosystems) technology according to the manufacturer’s instructions, on an ABI PRISM 3700 (Appied Biosystems). Sequencing was carried out from the 5′ or the 3′ end. Base calling and trimming of low-quality sequences was done using Phred. Sequences that were shorter than 100 bp (excluding from polyA tail) were not analyzed further. Sequence analysis was carried out using cross_match for vector masking. Contigs were built using the Phrap program. Database searches for E-values below 10−5 were used to infer a potential function or identify a putative contamination. Homology searches for function assignment were based on the results obtained with the SwissProt comparison. For the contigs and singletons for which no matches were found, TrEMBL comparisons were made. ESTs composed of regions that matched different proteins were classified as chimeric. Mitochondrial and plastid sequences were identified using the BLASTN program on Arabidopsis sequences.
Virtual Northern analysis
A 1 µl aliquot of a plasmid miniprep was amplified by PCR using M13 forward and M13 reverse primers in reaction volumes of 25 µl. Amplifications were performed in a GeneAmp PCR system 9700 cycler (Perkin Elmer). PCR mixes contained 2.5 µl of 10× reaction buffer (200 mM TRIS-HCl pH 8.4, 500 mM KCl), 2.5 mM MgCl2, 200 µM dNTPs, each primer at 0.2 µM, and 0.5 U of Taq DNA Polymerase (Life Technologies). An initial 5-min denaturation step was followed by 20 cycles of 30 s at 94°C, 30 s of annealing at 52°C, and a 2-min elongation at 72°C. One microliter of each PCR product was analyzed on by electrophoresis on a 1% agarose gel, transferred to nylon filters and hybridized with radiolabelled probes corresponding to SMART cDNAs from each condition; these cDNAs were prepared by random priming.
Filters were pre-hybridized and hybridized at 65°C in 5×SSC, 0.1% SDS, 1% powdered milk and salmon sperm DNA (100 ng/ml). Filters were washed twice for 30 min each at room temperature in 2×SSC, for 30 min at 60°C in 2×SSC/0.1% SDS and for 30 min at room temperature in 0.1×SSC. Two successive film exposures of 16 h and 10 days were performed.
Results
When sunflower protoplasts are cultured in liquid medium, they divide symmetrically, giving rise to loose colonies which define the non-embryogenic condition. When protoplasts are cultured embedded in agarose, they divide asymmetrically and follow a developmental pattern similar to that of the fertilized zygote, giving rise to so-called embryoids which define the embryogenic condition. In order to identify the genes expressed during the asymmetrical division event, four different cDNA libraries were constructed and an arbitrary fraction of each library was sequenced. Culture time was limited to 5 days in each case, because the first division takes place during this period in liquid and in solid media (Petitprez et al. 1995).
Protoplasts developing in non-embryogenic conditions: the HaDplR2 library
HaDplR2 (H elianthus a nnuus Development of protoplasts in liquid medium, Reference library number2) is a reference cDNA library constructed from mRNA isolated from protoplasts cultured for between 1 and 5 days under the non-embryogenic condition. The efficiency of transformation was 1.2×106 transformants/μg of cDNA. All of the selectable clones obtained from 31 ng of cDNA were arrayed, resulting in 9984 arranged clones. In all, 1632 cDNA clones were sequenced and 743 ESTs have been submitted to the EMBL database (Accession Nos. AJ412352–AJ412520, AJ412521–AJ412667, and AJ437699–AJ437975).
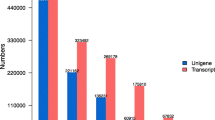
From the 1632 sequencing reactions, only 968 ESTs were obtained. The average length of sequence of good quality was 407 bp and the average EST length 398.9 bp (Table 1). After quality trimming, 805 sequences remained (two putative E. coli sequences were discarded).
A total of 118 contigs, made up of 335 ESTs, were generated using the Phrap program: 89 contigs result from the assembly of two sequences each, the remainder comprise less than ten sequences each, with the exception of the major contig, which is composed of 57 sequences. According to the Phrap analysis, contigs and singletons define 588 different sequences (Table 2).
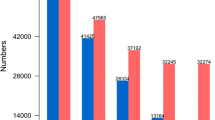
In order to attribute a putative function to the products encoded by the genes represented by the ESTs, public databases were searched at E-values below 10−5. Function assignment was performed based on results of comparisons with SwissProt, or with TrEMBL when no homologies were found with the former. The BLASTX program allowed us to attribute a putative function to 56% of contigs and singletons. According to the results of the BLAST analysis, sequences were classified into eighteen groups based on the KEGG catalog (Fig. 1). The major group was related to translation functions (11.4%). Some 34.6% of ESTs showed no match to any known protein, and these sequences may represent novel genes. This value may be an overestimate, as 38% of these ESTs were obtained from the 3′-end and many of those that do not find a homologous sequence in a protein database might be derived from 3′-UTRs.

Graphical representation of EST frequencies in each of the four libraries, based on the functional classification in the Kyoto Encyclopedia of Genes and Genomes (KEGG)
Protoplasts developing under embryogenic conditions: the HaDpsR1 library
HaDpsR1 (H. a nnuus Development of protoplasts in solid medium, Reference library number1) is a cDNA reference library constructed with mRNA from protoplasts cultured for between 1 and 5 days under the embryogenic condition. The primary cDNA library, constructed in pTriplEX2, had a titre of 1011 pfu, indicating adequate representation of the original mRNA pool. Of the 5404 cDNA clones arrayed, 1056 were sequenced. In all, 741 EST sequences remained after quality trimming and were submitted to the EMBL database (Accession Nos. AJ541055–AJ541795). The average length of good-quality sequence was 430 bp, and the average EST length was 310 bp (Table 1). Phrap assembled 379 sequences into 82 contigs, the major one containing 127 sequences. A total of 55 contigs are each composed by only two sequences. A total of 444 unique sequences were obtained from the sequenced ESTs according to Phrap results (Table 2).
BLASTX searches in public databases permitted us to attribute a putative function to 43% of the sequences. As in the case of HaDplR2, the results of sequence homology searches were used to classify putative proteins encoded by contigs and singletons with respect to eighteen different functional groups (Fig. 1). The most important groups are related to stress (11.1%) and protein synthesis (9.4%). Some 31.8% of sequences do not match any of the proteins in databases.
Enrichment for ESTs from protoplasts cultured under embryogenic conditions: the HaSemS3 library
HaSemS3 (H. a nnuus Somatic embryogenesis, Subtractive library number3) is a subtractive cDNA library enriched in transcripts expressed in protoplasts cultured under embryogenic conditions. This library is composed of 3072 cDNA clones obtained by plating all of a secondary round of PCR amplification. A total of 1056 clones were sequenced, and 644 EST sequences remained after quality trimming and were submitted to the EMBL database (Accession Nos.: AJ539583–AJ540226). Average good-quality sequence length is 384 bp and the average EST is 294 bp long (Table 1).
The Phrap program assembled 293 sequences into 63 contigs: the major contig was composed of 33 sequences, and 57 other contigs are made up of ten sequences or less. According to the Phrap results, 414 unique sequences were obtained (Table 2).
Similarity searches using BLASTX comparisons allowed attribution of a putative function to 63% of contigs and singletons. Different ESTs were classified into 18 different groups based on BLASTX similarities. The largest group consisted of genes predicted to have a function in defense or stress responses (28%) (Fig. 1). Some 29.1% of sequences do not match any known protein.
Enrichment for ESTs from protoplasts cultured under non-embryogenic conditions: the HaSemS4 library
The HaSemS4 (H. a nnuus Somatic embryogenesis, Subtractive library number4) library is the complementary library to HaSemS3, as it is enriched in transcripts that are preferentially expressed in protoplasts cultured under non-embryogenic condition. This library is made up of 4416 cDNA clones, of which 1056 were sequenced. Average good quality sequence length was 449 bp and the average EST length was 294 bp. In all, 292 ESTs were deposited in the EMBL database (Accession Nos.: AJ542101–AJ542392). After quality trimming only 289 sequences remained (putative E. coli sequences were discarded, see Table 1). This low rate is explained by the short length of ESTs after vector and quality trimming, many of them being removed from the analysis because they were less than 100 bp in length (excluding the polyA tail). The Phrap program assembled 99 sequences into 31 contigs: the largest of which was composed by 13 sequences. According to the Phrap results, 221 unique sequences were obtained (Table 2). Based on BLASTX comparisons, 64% of the unique sequences showed a significant match with one or more entries in TrEMBL or SwissProt databases, but a putative function could be attributed to only 52% of the sequences. Contigs and singletons were classified into 18 groups based on their predicted function; the largest groups consisted of genes predicted to encode proteins associated with gene expression and protein synthesis (i.e. transcription and translation; 10%) or to participate in primary metabolism (i.e. carbohydrate, energy, lipid and fatty acid, and amino acid metabolism; 15%) (Fig. 1). Some 36.3% of ESTs do not show homology to any protein in databases.
Assessment of the efficiency of subtraction by virtual Northern analysis
In order to assess subtraction efficiency in both subtractive libraries, virtual Northern blot analysis were carried out. A total of 192 PCR-amplified cDNA inserts from each library were resolved in duplicate on high-density gels, and immobilized on nylon membranes by Southern blotting. Duplicate filters were hybridized with double-stranded cDNAs obtained using the SMART technology after reverse transcription of mRNA from protoplasts cultured under both conditions. SMART technology was necessary since the cell culture conditions do not allow us to obtain sufficient mRNA for conventional probe preparation. The same number of cells, the same quantity of RNA and identical procedures for SMART amplification procedure, labeling and autoradiography were used for the comparison of HaSemS3 and HaSemS4 libraries. Filters were subjected to two successive exposures for film autoradiography, the first exposure for overnight and the second exposure for 10 days (Fig. 2).

Example of Virtual Northern on 96 HaSemS3 clones after a 10-day exposure. a Hybridization with cDNA from protoplasts cultured under non-embryogenic conditions. b Hybridization with cDNA from protoplasts kept under embryogenic conditions
Table 3 summarizes the results. For HaSemS3, 10.4% of clones showed an ON/OFF expression pattern and 24% displayed differential expression under the two conditions after overnight exposure; these clones thus represent abundant transcripts. Some 50% of the clones showed no signal. After a 10-day exposure, 25% exhibit no signal at all, indicating that this library is enriched in rare transcripts.
Similarly, 25% of the clones from the HaSemS4 library showed an ON/OFF pattern and 49% were differentially expressed based on the signal patterns after overnight exposure. Since 12% of clones gave no signal after overnight exposure, but only 7.3% failed to detect transcripts after 10 days, we conclude that this library is less enriched in rare transcripts than is HaSemS3.
Discussion
The main aim of the present work was to obtain an initial overview of the genes expressed in protoplasts under embryogenic and non-embryogenic conditions, with the latter serving as a model for the first asymmetrical division of the fertilized zygote. Assuming that 10% of genes are expressed under each condition, one would expect approximately 3000 different transcripts. Combining the analysis of 1000 clones from a reference library and 1000 clones from a subtractive library would thus allow us to access the most highly expressed (from the reference library) and some less expressed genes (from the subtractive libraries).
The genetic program expressed under non-embryogenic conditions
Many regulatory processes occur post-transcriptionally. However clues to which types of functions are of most importance in any particular tissue or condition can be inferred from the general distribution of ESTs among different functional groups, as shown in Fig. 1. Having constructed a reference library, we hypothesized that the number of ESTs corresponding to a particular contig should reflect the abundance of the corresponding transcript in the tissue analyzed. The reliability of this information is proportional to the number of ESTs present in the contig. The analysis of a subtracted library complements this information, because it gives access to less expressed genes. From the results obtained with the Phrap assembly program for the reference library (Table 2), we deduce that HaDplR2 is a library characterized by low redundancy, because there are on average 2.8 ESTs per contig and only one transcript is highly represented.
Comparison of the HaDplR2 and HaSemS4 libraries showed that protoplasts cultured under non-embryogenic conditions mainly expressed genes encoding proteins related to protein synthesis (10.5% and 9%, respectively) and primary metabolism (6.6% and 5.5%, respectively, for amino acid metabolism and 6.9% for energy metabolism in HaSemS4) (Fig. 1). This is consistent with the expected cellular functions of protoplasts growing in liquid medium; such cells divide to form colonies, and may thus be assumed to have a simple vegetative activity. The most abundant group of ESTs was found to code for products that are involved in translation of mRNAs.
Table 4 summarizes for each library the number of sequences that display homology with sequences in different databases. For the HaDplR2 and HaSemS4 libraries, more than 50% of ESTs (56.3% and 52.9% for HaDplR2 and HaSemS4, respectively) showed no homology with previously described sunflower ESTs, making these cDNA resources quite UNIQUE among the 47,000 published sunflower ESTs. Interestingly, in both libraries more than 25% of the sequences were not homologous to any plant ESTs or proteins; thus, these represent putatively sunflower-specific sequences or sequences expressed in physiological situations or tissues that were less extensively sampled in other plant EST programs.
The genetic program expressed under embryogenic conditions
Comparison of HaDpsR1 and HaSemS3 showed that stress-related genes are highly represented in both libraries (Fig. 1). This result was expected in light of the conditions encountered by protoplasts during culture in solid medium: the cells are exposed to the heat shock associated with embedding in agarose and are probably highly stressed. Beside the stress-related group, the most abundant transcripts (estimated on the basis of EST frequencies) were found in the translation-related group. However, in the HaDpsR1 library, the translation-related group is slightly smaller than the stress-related group, whereas stress-related proteins are three times more abundant than translation related proteins in the HaSemS3 library. Thus, the relative increase in stress-related transcripts in HaSemS3 may be a consequence of a less-efficient normalization step. The abundance of genes of unknown function, with one highly represented transcript (127 ESTs in the contig), expressed under embryogenic conditions may reflect the presence of some interesting but yet unstudied genes having a key function in protoplasts cultured under this condition.
The results in Table 4 show that a large percentage of previously unknown sunflower sequences is in both libraries (44.6% for HaDpsR1 and 35.1% for HaSemS3). However, these figures are lower than those for the libraries representing cells cultured in non-embryogenic conditions. Some 15% of ESTs from HaDpsR1 and 25% from HaSemS3 correspond to transcripts with no homology to other known proteins or transcripts from dicots (Table 4, category 8).
Comparison between embryogenic and non-embryogenic libraries
The method proposed by Stekel et al. (2000) for comparing gene expression profiles from multiple cDNA libraries was used to assess the degree of difference in expression between ESTs from HaDplR2 and HaDpsR1. Six contigs with an R statistic >6 were identified; the R statistic estimates the extent to which the difference in gene expression corresponds to the heterogeneity of the libraries. The top hits are shown in Table 5, with a brief description of the protein and the counts of the gene in each library. The list contains a ribosomal protein that is differentially expressed under the two conditions studied here. This result is consistent with previously reported data which suggested that ribosomal genes appear to be differentially regulated between different tissue types (Karsi et al. 2002). We also identified an rDNA promoter binding protein which may control ribosomal protein expression. Another differentially expressed gene codes for a trypsin inhibitor, a class of protein known to be expressed in stressed tissues. As protoplasts that are maintained under embryogenic conditions are highly stressed, its expression under these conditions is expected. Among the contigs that show a significant level of differential expression, several present homology with hypothetical proteins of unknown function. Some of these genes may respond to stress or differences in culture conditions, while others may encode very important proteins involved in the establishment of asymmetric division. The latter are currently under investigation.
Subtractive libraries are highly enriched in differentially expressed transcripts
The comparison of subtractive libraries should help to identify genes involved in the acquisition of polarity in the early embryo. HaSemS4 is the complementary library of HaSemS3, so one could expect a low level of EST redundancy of between the libraries. Subtraction efficiency is usually estimated by Virtual Northern hybridization using marker genes known to be differentially expressed. This experiment could not be performed in our case because there is as yet no clear marker gene for the studied conditions. A PHRAP assembly was performed using ESTs from all libraries; only 27 contigs made up of ESTs from both subtractive libraries were identified, indicating that subtraction efficiency was quite high. Another important issue is the low rate of ESTs from constitutively expressed genes such as EF1α, which was found twice in HaDpsR1 and five times in HaDplR2.
The Virtual Northern blots carried out on the subtractive libraries showed that both libraries are enriched in differentially expressed genes, as they present a high percentage of transcripts with a differential expression pattern (see Table 3). Data from PHRAP assembly showed that 87% of contigs and singletons from HaSemS3 are differentially expressed between the two conditions versus 84% from HaDpsR1; i.e., HaSemS3 exhibits a slightly higher proportion of differentially expressed genes. However, HaSemS4 comprises 83% differentially expressed transcripts versus 88% of contigs and singletons in HaDplR2. The number of differentially expressed genes in HaDplR2 may be an overestimate; PHRAP might be unable to assemble ESTs obtained from the 5′- and the 3′- ends of the same gene.
Stress related transcripts are highly represented in HaSemS3 (28% versus 6.2%) while in HaSemS4 the proportion of ESTS related to primary metabolism is higher (15% versus 6%, Fig. 1). One notable difference between the libraries is the percentage of sequences having a putative function. In HaSemS4, this percentage is lower than in HaSemS3 (52% versus 63.5%). This difference could be a result of the generally lower quality of the HaSemS4 sequences.
We have already identified some key genes based on their homology with animal and plant genes involved in asymmetrical zygote division. For example, mago nashi -like sequences (Accession Nos. AJ541081, AJ541776 and AJ540176) were found in the libraries constructed from protoplasts cultured under embryogenic conditions (HaDpsR1 and HaSemS3) with E value below 10−70. The mago nashi gene was first described in Drosophila melanogaster, where it plays a role in axis formation during oogenesis (Boswell et al. 1991). A mago nashi ortholog has also been described in C. elegans, which functionally complements the D. melanogaster mutant. The MAGO NASHI protein is highly conserved among species and homologs have been described in yeast and other fungi as well as in plants. The involvement of this protein in polarity acquisition in sunflower protoplasts is under investigation. Interestingly, several putative RNA-binding proteins, considered to be putative partners of the mago nashi product, are also present in every library. A sunflower homolog of the Drosophila gene Argonaute-1 (Accession No. AJ542157, E=10−19) was also identified. Argonaute-1 is implicated in post-transcriptional gene silencing and plays an essential role in neural growth in Drosophila (Williams and Rubin 2002). The plant homolog ago1 has been reported to act in the establishment of bilateral embryo symmetry in A. thaliana (Lynn et al. 1999; Carmell et al. 2002). Intriguingly, the homology to ago1 was found in HaSemS4. Members of the family of SHAGGY-like kinases, which are involved in different processes of flower development and brassinosteroid signaling (Jonak and Hirt 2002), were also identified in HaDplR2 and HaSemS3.
The data presented here provide the first overview of genes expressed in sunflower protoplasts cultured under embryogenic and non-embryogenic conditions, and our analysis of the cDNA libraries represents progress towards a better understanding of the genetic basis of symmetrical and asymmetrical cell divisions in this crop plant. A total of 4800 ESTs were sequenced. After quality trimming, these ESTs were found to represent a unigene set composed by 1502 sequences. This analysis allowed us to identify 821 previously uncharacterized sunflower sequences, and significant differences in expression of transcripts between embryogenic and non-embryogenic conditions were deduced from EST frequencies. We also identified sunflower homologs of key genes involved in animal and plant embryo development. The characterization of the genes implicated in embryo polarity establishment will be completed by functional genomic approaches using these libraries to construct cDNA microarrays. The libraries represent a valuable resource for discovering genes potentially involved in induction or determination of embryo polarity in dicot plants, and could also be useful for resource for developmental genomic studies on exalbuminous dicots.
References
Alibert G, Aslane-Chanabe C, Burrus M (1994) Sunflower tissue and cell cultures and their use in biotechnology. Plant Physiol Biochem 32:31–44
Belanger K, Quatrano R (2000) Polarity: the role of localized secretion. Curr Opin Plant Biol 3:67–72
Boswell RE, Prout ME, Steichen JC (1991) Mutations in a newly identified Drosophila melanogaster gene, mago nashi , disrupt germ cell formation and result in the formation of mirror-image symmetrical double abdomen embryos. Development 113:373–384
Busch M, Mayer U, Jürgens G (1996) Molecular analysis of the Arabidopsis pattern formation gene GNOM: gene structure and intragenic complementation. Mol Gen Genet 250:681–691
Caboche M, Boucly M (2000) The Genoplante programme, a mobilizing programme in plant genomics. Comptes Rend Acad Agric France 86:159–173
Carmell MA, Xuan Z, Zhang MQ, Hannon GJ (2002) The Argonaute family: tentacles that reach into RNAi, developmental control, stem cell maintenance, and tumorigenesis. Genes Dev 16:2733–2742
Chanabé C, Burrus M, Alibert G (1989) Factors affecting the improvement of colony formation from sunflower protoplasts. Plant Sci 64:125–132
Chanabé C, Burrus M, Bidney D, Alibert G (1991) Studies on plant regeneration from protoplasts in the genus Helianthus. Plant Cell Rep 9:635–638
Chaudhury A, Koltunow A, Payne T, Luo M, Tucker M, Dennis E, Peacock W (2001) Control of early seed development. Annu Rev Cell Dev Biol 17:677–699
Chen J, Ullah H, Young J, Sussman M, Jones A (2001) ABP1 is required for organized cell elongation and division in Arabidopsis embryogenesis. Genes Dev 15:902–911
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159
Flores Berrios E, Gentzbittel L Kayyal H, Alibert G, Sarrafi A (2000) AFLP mapping of QTLs for in vitro organogenesis traits using recombinant inbred lines in sunflower ( Helianthus annuus L.). Theor Appl Genet 101:1299–1306
Gentzbittel L, Abbott A, Galaud J, Georgi L, Fabre F, Liboz T, Alibert G (2002) A bacterial artificial chromosome (BAC) library for sunflower, and identification of clones containing genes for putative transmembrane receptors. Mol Genet Genomics 266:979–987
Goldberg R, de Paiva G, Yadegari R (1994) Plant embryogenesis: zygote to seed. Science 266:605–614
Hardtke C, Berleth T (1998) The Arabidopsis gene MONOPTEROS encodes a transcription factor mediating embryo axis formation and vascular development. EMBO J 17:1405–1411
Jonak C, Hirt H (2002) Glycogen synthase 3/SHAGGY-like kinases in plant: an emerging family with novel functions. Trends Plant Sci 7:457–461
Jürgens G (1995) Axis formation in plant embryogenesis: cues and clues. Cell 81:467–470
Jürgens G (2001) Apical-basal pattern formation in Arabidopsis embryogenesis. EMBO J 20:3609–3616
Jürgens G, Grebe M, Steinmann T (1997) Establishment of cell polarity during early plant development. Curr Opin Cell Biol 9:849–852
Karsi A, Patterson A, Feng J, Liu Z (2002) Translational machinery of channel catfish: I. A transcriptomic approach to the analysis of 32 40S ribosomal protein genes and their expression. Gene 291:177–186
Lynn K, Fernandez A, Aida M, Sedbrook J, Tasaka M, Masson P, Barton MK (1999) The PINHEAD/ZWILLE gene acts pleiotropically in Arabidopsis development and has overlapping functions with the ARGONAUTE1 gene. Development 126:469–481
Mayer U, Büttner G, Jürgens G (1993) Apical-basal pattern formation in the Arabidopsis embryo: studies on the role of the GNOM gene. Development 117:149–162
Meinke DW (1985) Embryo-lethal mutants of Arabidopsis thaliana: analysis of mutants with a wide range of lethal phases. Theor Appl Genet 69:543–552
Petitprez M, Briere C, Borin C, Kallerhoff J, Souvre A, Alibert G (1995) Characterisation of protoplasts from hypocotyls of Helianthus annuus in relation to their tissue origin. Plant Cell Tissue Org Cult 41:33–40
Robertson N, Khetarpal U, Gutierrez-Espeleta G, Bieber F, Morton C (1994) Isolation of novel and known genes from a human fetal cochlear cDNA library using subtractive hybridization and differential screening. Genomics 23:42–50
Souter M, Lindsey K (2000) Polarity and signalling in plant embryogenesis. J Exp Bot 51:971–983
Stekel D, Git Y, Falciani F (2000) The comparison of gene expression from multiple cDNA libraries. Genome Res 10:2055–2061
Tang S, Yu JK, Slabaugh B, Shintani K, Knapp J (2002) Simple sequence repeat map of the sunflower genome. Theor Appl Genet 105:1124–1136
Torres-Ruiz R, Jürgens G (1994) Mutations in the FASS gene uncouple pattern formation and morphogenesis in Arabidopsis development. Development 120:2967–2978
West M, Harada JJ (1993) Embryogenesis in higher plants: an overview. Plant Cell 5:1361–1369
Williams RW, Rubin GM (2002) ARGONAUTE1 is required for efficient RNA interference in Drosophila embryos. Proc Natl Acad Sci USA 99:6889–6894
Zimmerman JL (1993) Somatic embryogenesis: a model for early development in higher plants. Plant Cell 5:1411–1423
Acknowledgments
We thank Nathalie Ladouce for her technical support, Philippe Anson for protoplast production and Sébastien Moretti for assistance in EST annotation. This work was supported by grants from the Toulouse Genomics Center (French Network of Genomics Centers) and the Conseil Régional de Midi-Pyrénées (programme Génomique et PostGenomique). CT is supported by a doctoral fellowship of the French government
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Hagemann
Rights and permissions
About this article
Cite this article
Tamborindeguy, C., Ben, C., Liboz, T. et al. Sequence evaluation of four specific cDNA libraries for developmental genomics of sunflower. Mol Genet Genomics 271, 367–375 (2004). https://doi.org/10.1007/s00438-004-0989-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-004-0989-5