
Abstract
Western white pine (Pinus monticola Dougl. ex. D. Don., WWP) shows genetic variation in disease resistance to white pine blister rust (Cronartium ribicola). Most plant disease resistance (R) genes encode proteins that belong to a superfamily with nucleotide-binding site domains (NBS) and C-terminal leucine-rich repeats (LRR). In this work a PCR strategy was used to clone R gene analogs (RGAs) from WWP using oligonucleotide primers based on the conserved sequence motifs in the NBS domain of angiosperm NBS-LRR genes. Sixty-seven NBS sequences were cloned from disease-resistant trees. BLAST searches in GenBank revealed that they shared significant identity to well-characterized R genes from angiosperms, including L and M genes from flax, the tobacco N gene and the soybean gene LM6. Sequence alignments revealed that the RGAs from WWP contained the conserved motifs identified in angiosperm NBS domains, especially those motifs specific for TIR-NBS-LRR proteins. Phylogenic analysis of plant R genes and RGAs indicated that all cloned WWP RGAs can be grouped into one major branch together with well-known R proteins carrying a TIR domain, suggesting they belong to the subfamily of TIR-NBS-LRR genes. In one phylogenic tree, WWP RGAs were further subdivided into fourteen clusters with an amino acid sequence identity threshold of 75%. cDNA cloning and RT-PCR analysis with gene-specific primers demonstrated that members of 10 of the 14 RGA classes were expressed in foliage tissues, suggesting that a large and diverse NBS-LRR gene family may be functional in conifers. These results provide evidence for the hypothesis that conifer RGAs share a common origin with R genes from angiosperms, and some of them may play important roles in defense mechanisms that confer disease resistance in western white pine. Ratios of non-synonymous to synonymous nucleotide substitutions (Ka/Ks) in the WWP NBS domains were greater than 1 or close to 1, indicating that diversifying selection and/or neutral selection operate on the NBS domains of the WWP RGA family.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Western white pine (Pinus monticola Dougl.) is an important species of the forest ecosystem in western North America. White pine blister rust (Cronartium ribicola J.C. Fisch.) has caused widespread mortality in western white pine (WWP) and other five-needle pines since it was introduced to North America around 1910. The rust has had a severe impact on forest ecosystems and has resulted in reduced planting of P. monticola and other five-needle pines. Resistance to C. ribicola and maintenance of genetic diversity in production populations are very important for WWP breeding. Currently genetic resistance is the key management tool that provides an approach to re-establishing five-needle pines in North America. In western white pine, several types of resistance to white pine blister rust have been identified, involving, e.g., slow canker growth (SCG) (Hunt 1997) and dominant disease resistance genes (Cr2) (Kinloch et al. 1999). These resistance resources are currently being used in white pine breeding programs in British Columbia (BC). An SCG resistance is expressed in the stems of western white pine seedlings that restricts infection area, and sometimes the canker heals over. Different types of SCG have been observed, suggesting that this disease resistance may be controlled by multiple genes (Ekramoddoullah and Hunt 2002). On the other hand, Cr2 -mediated resistance is a qualitative trait and occurs in the needles, setting off a classical hypersensitive reaction (HR), preventing stem infection by C. ribicola.
Plant disease-resistance genes (R genes) are postulated to encode receptors for the specific recognition of avirulence gene (Avr) products synthesized by the pathogen. The interaction of R genes and avirulence genes from pathogens is presumed to trigger a network of signal-transduction cascades that result in a series of plant defense processes, including the induction of pathogenesis-related (PR) proteins, which ultimately lead to disease resistance (Innes 1998). In recent years, several approaches have been adopted to investigate plant defense mechanisms in conifer pathosystems during fungal infection (Ekramoddoullah and Hunt 2002). An analysis of protein profiles was used to search for differentially expressed proteins, including some PR proteins, from resistant and susceptible conifer seedlings during pathogen infection (Ekramoddoullah and Hunt 1993; Ekramoddoullah et al.1998). A significantly increased level of PR10 protein was observed in needles of resistant sugar pine (carrying the dominant resistance gene Cr1) following infection by C. ribicola (Ekramoddoullah and Tan 1998). A more detailed investigation revealed that multiple PR10 proteins and genes are differentially expressed in WWP in response to fungal infection, wounding and low temperatures (Liu et al. 2003). Immunocytochemical analysis demonstrated the localization of PR10 protein around the cells of the blister rust fungus C. ribicola in infected spots of the susceptible WWP needles. PR10 protein was also found, but the pathogen itself was no longer detectable in the hypersensitive infection spots on sugar pine. Histochemical examination revealed an extra layer of endodermal cells surrounding the vascular bundle at the hypersensitive infection spots in the white pine needles (Ekramoddoullah and Hunt 2002). This type of physical barrier induced by infection is proposed to contribute to rust resistance in conifers. Other PR proteins such as endochitinases, thaumatin-like proteins, and a putative anti-fungal protein have also been identified in conifers following fungal infection (Davidson and Ekramoddoullah 1997, Robinson et al. 2000, and unpublished data).
Over 30 disease resistance genes have now been cloned from model and crop species of angiosperms; they confer resistance to various plant pathogens, including viruses, bacteria, oomycetes, fungi, nematodes and insects (Ellis and Jones 1998, Ferrier-Cana et al. 2003). R genes are grouped into five types based on the structure of their protein products (Dangl and Jones 2001). Most R genes encode proteins containing a nucleotide-binding site (NBS) domain and a stretch of leucine-rich repeats (LRR) at the C-terminus. Other R proteins include those with only an LRR domain and no NBS domain, an LRR domain and a serine-threonine protein kinase domain without a NBS domain, a protein kinase domain without an NBS or an LRR domain, or a coiled-coil (CC) domain plus a putative signal anchor at the N-terminus. Based on motifs located N-terminal to the NBS domain, the NBS-LRR superfamily can be further subdivided into the TIR (Drosophila Toll and mammalian interleukin-1 receptor homology regions) subfamily and the non-TIR subfamily, members of which typically contain N-terminal CC domains (Meyers et al. 1999; Pan et al. 2000a; Young 2000; Cannon et al. 2002). The NBS-LRR genes are abundant in plants. A total of 166 NBS-LRR genes are present in the genome of Arabidopsis thaliana ecotype Col-0, 70% of which belong to the TIR-NBS-LRR subfamily (Richly et al. 2002).
Eight conserved motifs have been identified in the NBS domains (Meyers et al. 1999), some of which are specific to the non-TIR or the TIR-NBS-LRR subfamily. These motifs have been used to synthesize degenerate primers for the isolation of disease resistance gene analogues (RGAs) containing NBS sequences from both dicot and cereal genomes (Cannon et al. 2002). This PCR cloning strategy has been employed to clone RGA sequences from soybean (Kanazin et al. 1996, Yu et al. 1996, Peñuela et al. 2002), potato (Leister et al. 1996), lettuce (Shen et al. 1998), bean (Creusot et al. 1999, Rivkin et al. 1999), Arabidopsis (Aarts et al. 1998, Speulman et al. 1998), maize (Collins et al. 1998), rice and barley (Leister et al. 1998), and other angiosperm species.
Our long-term objective is to isolate the Cr2 gene, which confers resistance against C. ribicola to western white pine. This has prompted us to clone RGAs from this gymnosperm species. In the present study, we used a PCR cloning strategy with degenerate primers to successfully amplify NBS sequences from the white pine genome. Sequence determinations and phylogenetic analyses were performed to elucidate their relationships with well-known R genes in angiosperms. The RGAs from western white pine were investigated for their diversity and expression, in order to gain insights into the evolution of TIR-NBS-LRR genes and their potential for use in disease resistance breeding.
Materials and methods
Plant material
Western white pine seed lots #3277 and #3278, both known to contain the Cr2 gene, were obtained from Dorena, USA (Kinloch et al. 1999; Registration Nos. 119-15045-845 X OP and 119-15045-845 X 15045-841 X OP), sown and grown at the Pacific Forestry Centre in Victoria (British Columbia) in 1994. They were inoculated in the ribes (disease) garden. Survivors were confirmed as resistant by re-inoculation the following year. Needles from these survivors were collected in 2001 for the isolation of genomic DNA and total RNA.
PCR and molecular cloning
Six oligonucleotide primers were designed for PCR cloning of RGA sequences from western white pine. These primers targeted three highly conserved motifs (P-loop/kinase-1, Kinase-2, GLPL) and one less conserved motif (DMGRDL) in the NBS domain of well-characterized resistance genes from angiosperm plants (Table 1). Because highly degenerate primer sets, such as Gkt2/Pal2, amplified a high proportion of non-specific fragments of about the same sizes as the RGAs, the primers Dvd1, Pal1 and Dmg1 were designed as unique sequences to avoid non-specific amplification (Table 1). A nested primer strategy was applied in some PCR experiments using primers for more than two conserved motifs. In the experiments with the nested primer approach, the diluted first-round PCR products were used as templates for the second-round PCR with every possible primer combination for two internal motifs, or for one internal motif and one motif targeted in the first-round PCR.
Genomic DNA was prepared from current-year needles of individual seedlings using a DNeasy kit (Qiagen, Mississauga, Ont., Canada). PCR was performed using a PCR Master Mix Kit (Qiagen) in a final volume of 50 μl, containing 100 ng of genomic DNA on a Perkin-Elmer Thermocycler (Perkin-Elmer Applied Biosystems, Foster City, Calif. USA). Thermal cycling conditions consisted of an initial denaturation step at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 30 s, primer annealing at 42°C for 1 min and primer extension at 72°C for 1 min, with a final 10-min extension at 72°C.
After agarose gel fractionation, the amplified DNA fragments of expected sizes (assuming no introns) were excised and purified using a MinElute gel extraction kit (Qiagen) and cloned into the pGEM-T easy vector (Promega, Madison, Wis., USA). Selection and manipulation of recombinant plasmid clones followed standard methods (Sambrook et al. 1989). To discriminate between different sequences derived from PCR, multiple clones were analyzed by restriction enzyme digestion with Taq I or Rsa I, and grouped according to their digestion patterns. For each group, at least two clones were picked for DNA sequence analysis. In most cases, the two clones from a given group had the same sequence. If not, more clones from these groups were analyzed to confirm DNA sequences and identify possible errors that had resulted from misincorporation during PCR.
DNA sequence analysis
DNA sequences of RGA clones were determined on both strands using an ABI310 DNA sequencer (Applied Biosystems), a Thermo-cycle sequence kit (Amersham), and T7, SP6 and other internal primers as needed, according to the manufacturer’s instructions. DNA sequence data were compiled and analyzed using BLAST (Altschul et al. 1997) and ClustalW network services at the National Center for Biotechnology Information (NCBI). A phylogenetic tree based on the NBS sequence alignment was constructed by the Fitch-Margoliash and least-squares methods with evolutionary clock (Kuhner and Felsenstein 1994) using the BioEdit package (Hall 1999). The reliability of the tree was established by conducting 1000 neighbor-joining bootstrap sampling steps. The RGA sequences from western white pine have been deposited in the GenBank database under the Accession Nos. AY294058–AY294124.
cDNA preparation and amplification
RNA was isolated from pooled foliage of multiple rust-resistant trees using the procedure described previously (Liu and Ekramoddoullah 2003). Total RNA was treated with RQ 1 RNase-Free DNase (Promega) and purified again using a Plant RNeasy extraction kit (Qiagen). cDNA was reverse-transcribed using a SMART cDNA library construction kit (Clontech Laboratories, Palo Alto, Calif., USA) following the manufacturer’s instructions. PCR was carried out with 1 μl of cDNA in 25 μl of PCR mix using Taq PCR Master Mixture (Qiagen). The procedure for RT-PCR cloning of RGAs was the same as that used for RGA cloning from genomic DNA, except that cDNA was used as the template for PCR.
To examine the expression of RGAs cloned from genomic DNA, RT-PCR was performed with 18 pairs of RGA-specific primers based on the analysis of RGA nucleotide sequence alignments (Table 2). RT-PCR was carried out with 1 μl of cDNA in 25 μl of PCR mix using Taq PCR Master Mixture (Qiagen). Each RT-PCR was performed with negative controls (DNA and cDNA-free) and pooled genomic DNA from 10 resistant trees. The PCR products from templates of cDNA and genomic DNA were compared for the size and intensity. RGA expression levels were compared taking the genomic PCR product as reference. RT-PCR conditions were as described above for the NBS motif primers, except that the annealing temperature was 48°C and PCR was performed for 30 cycles.
Results
Analysis of WWP RGAs by PCR cloning
PCR using primer combinations corresponding to P-loop/DMG motifs yielded two major DNA bands of approximately 850 bp and 900 bp. PCRs using P-loop/GLPL, Kinase-2/DMG, or Kinase-2/GLPL primer pairs each produced only one DNA band of roughly 500 bp, 620 bp or 270 bp, respectively.
Sequence analysis revealed that only a small proportion of the clones obtained from the first-round PCR fragments displayed homology to R genes or RGAs of angiosperms. Only one out of 48 clones was identified as an RGA from the first-round of PCR with the primer combination Gkt2/Pal2. The nested primer strategy increased the specificity of the amplification of WWP RGAs. The second round of PCR with nested primers produced sharper DNA bands of the expected size assuming no introns. All sequenced clones from the nested PCR, including those performed with the primer sets Gkt1/Pal1, Dvd1/Dmg1 and Dvd1/Pal1, were RGAs, indicating that the nested PCR strategy was effective in increasing the efficiency of RGA cloning. In total 455 recombinant plasmids were identified with DNA inserts of expected sizes. Restriction enzyme analysis enabled the recombinant plasmids to be grouped into several classes. All unique groups were given a name based upon their cloning template and fragment length. At least two clones from each group were sequenced. BLAST searches led to the identification of 67 WWP RGAs, 21 from cDNA and 46 from genomic DNA (Table 3).
Analysis of RGA sequences revealed the presence of stop codon and/or frame shift mutations in 18 of 46 RGA genomic clones and in 14 of 21 RGA cDNA clones, while others contained an uninterrupted ORF (Fig. 1 and Table 3). Alignment of RGA nucleotide sequences showed that mutations were localized at the same position in several clones which were otherwise identical. The consistency of these RGA clones containing nonsense mutations therefore may not result from mistakes in PCR amplification or random errors in DNA sequence analysis, but may in fact indicate the presence of RGA pseudogene families in the WWP genome.

Alignment of the predicted amino acid sequences of NBS sequences from western white pine. Tobacco N (A54810) and flax L5 (AF093645) and M (AAB47618) are included in this analysis. Only 28 representative WWP RGAs that show ≤90% amino acid sequence identity are presented. Amino acid sequence identities are indicated by the dots. Premature stop codons and frame-shifts are indicated by asterisks and black triangles, respectively. The conserved motifs defined by Meyers et al. (1999) are highlighted
Comparison of NBS domain RGAs from WWP with angiosperm R genes
BLASTP searches of the GeneBank database demonstrated that the WWP RGAs shared significant homologies with well-characterized R genes from angiosperms, as indicated by the E values ranging from 4e-6 to 9e-44. Comparison of the deduced amino acid sequences of WWP RGAs with those predicted from R genes, such as tobacco N (A54810), flax M and L5 (AAB47618 and AF093645), and soybean LM6 (AAG09951), showed identities in the range of 24% to 36%. BLASTP searches also detected one homologous sequence from Pinus radiata (AF038649) and four others from P. taeda in GenBank (AY091558, AY091560, AY091562, and AY091566) (Meyers et al. 2002).
To establish the origin and elucidate the evolution of WWP RGAs, angiosperm R genes (AtRPS2, AtRPS5, BoRPS2, Lu-M, Lu-L, Nt-N) and other homologous conifer RGAs were included in this phylogenetic analysis. One phylogenetic tree was constructed from aligned sequences and contained two major branches (Fig. 2). As expected, AtRPS2, AtRPS5 and BoRPS2 were grouped together in one branch representing the non-TIR subfamily. All WWP RGAs identified in the current study were present in one branch containing the Nt-N, Lu-L5 , and Lu-M genes, suggesting that these WWP RGAs may belong to the TIR-NBS-LRR subfamily. This classification is further supported by the presence of internal conserved motifs in the NBS domain. Alignments of deduced amino acid sequences demonstrated that the conifer RGAs also contained the conserved motifs in NBS domains that were previously identified in R genes and RGAs of angiosperms (Meyers et al. 1999, Pan et al. 2000a, Peñuela et al. 2002), including the P-loop (Kinase-1), RNBS-A-TIR, Kinase-2, RNBS-b/Kinase-3, RNBS-C, GLPL, and RNBS-D-TIR signatures (Fig. 1). Among these conserved motifs, the P-loop and GLPL motifs are conserved in both TIR- and non-TIR-NBS-LRR proteins, while RNBS-A-TIR (LQKKLLSKLL) and RNBS-D-TIR (FLHIACFF) have been found only in the TIR-NBS-LRR proteins (Meyers et al. 1999). The discovery of a large number of RGAs in the WWP genome and their significant homologies with angiosperm TIR-NBS-LRR family provides evidence for the hypothesis that the TIR-NBS-LRR subfamily originated before the divergence of angiosperms and gymnosperms (Meyers et al. 1999, Pan et al. 2000a).

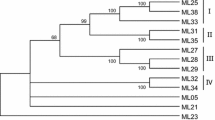
Phylogenetic tree based on the amino acid sequences of RGAs from western white pine and selected resistance proteins from angiosperms. RGA sequences from western white pine were compared to additional NBS-LRR type sequences with known disease resistance functions, including tobacco N (Nt-N), flax L5 (Lu-L5) and M (Lu-M), Arabidopsis AtRPS2 (AF487823) and AtRPS5 (AF074916), and Brassica BoRPS2 (AAF19803) proteins. Only 48 representative WWP RGAs that show ≤98% amino acid sequence identity are presented. The TIR-NBS-LRR branch is grouped into 14 classes of RGAs. Members of each class display ≤75% amino acid sequence identity with members of all other classes. The scale at the bottom indicates genetic distance proportional to the number of substitutions per site
Variation and evolutionary analysis of WWP RGA sequences
WWP RGAs are highly divergent, sharing 4–99% identity at the nucleotide level and 18–99% identity at the amino acid level in the NBS domain, indicating that, like angiosperms, conifers contain a large and diverse family of genes for proteins with the NBS domain. WWP RGAs appear as short-separated branches that form different clusters defined by levels of amino acid sequence identity ranging from 77% to 99% within clusters (Fig. 2). Fourteen clusters are defined at a sequence identity threshold of 75% (Fig. 2 and Table 3), with identity scores for pairwise comparisons of members from different classes ranging from 18% to 74%. Only one WWP RGA was identified in each of clusters 2, 6, 11, 13 and 14 (Table 3). RGA clone GI5033 was positioned at the base of the TIR-branch. It has higher identity to angiosperm R genes than to conifer RGAs. Its identity to TIR-type R or RGA sequences from both conifers and angiosperms ranges from 18% to 36%. This range is comparable to its identity (24% to 28%) to members of the non-TIR subfamily. This suggests that WWP RGAs might have arisen from a common ancestor of both TIR- and non-TIR gene families.
Thirty RGA cDNA clones obtained with primer sets based on Kin2 and MEG motifs were randomly picked for sequence analysis, yielding 18 different RGA sequences. They were divided into three types representing clones C601, C608 and C618, respectively (Table 3). RGAs of types C601 and C608 contained a continuous reading frame. However, RGA type C608, defined by 13 clones, had both premature stop codon mutations and frame-shift mutations. The reason for such a high rate of pseudogene recovery from a transcribed RGA type is not clear. C608-type RGAs were 99% homologous, which may in part reflect single nucleotide polymorphisms of allelic or paralogous pseudogenes in the WWP population, because the cDNA template was pooled from the foliage of multiple trees. However, several RGA genomic clones from a single seedling also showed nucleotide differences, suggesting that RGA gene duplication might have occurred in the WWP genome. Apart from single base differences, other types of mutations were also identified within the NBS domains. In 12 RGA clones of the C608 type a single nucleotide insertion resulted in a shift in the reading frame. A multiple base deletion at the same position was found in RGA clones C24, G6215, G8502 and G8533, which were grouped together in class 10 (Fig. 2).
To examine the evolutionary forces acting on NBS domains of WWP RGAs, the ratio of non-synonymous (Ka) to synonymous nucleotide substitutions (Ks) was calculated for members of five RGA classes (Table 4). The majority of nucleotide variations between RGA members represented non-synonymous substitutions. Ka/Ks values for pairwise comparisons ranged from 0.4 to ∞. One extreme example was the G6244/GM06 comparison, where every nucleotide change was nonsynonymous (Table 4). Ka/Ks ratios in many RGA comparisons were significantly larger than 1, while others showed no significant difference from the value expected for neutral selection (Ka/Ks=1) (Table 4). Pairwise comparisons involving the RGA pseudogene C608 did not change the pattern of Ka/Ks ratios. The divergent or neutral selection acting on the NBS domains of WWP RGAs shown by this analysis contrasts with the NBS domain of angiosperm RGAs, which are considered to be under conservative selection pressure (Michelmore and Meyers 1998, Graham et al. 2002).
Expression analysis of WWP RGAs
The expression profiles of selected RGAs are summarized in Table 3. RT-PCR showed that seven of 18 RGAs were expressed (Fig. 3). For the other 11 RGAs, the corresponding mRNAs were not detectable in foliage tissues. The control PCR produced the expected DNA fragments from the genomic DNA template using all 18 RGA-specific primer pairs. The different staining densities of RT-PCR products on agarose gels demonstrated that the expression levels of RGAs were different, because the same amount of cDNA template was added to each RT-PCR tube. As shown in Fig. 3, G9036 was expressed at the lowest level, G8106 and G6244 were expressed at the highest levels, and other three RGAs (G8029, G5004, and G8540) were expressed at intermediate levels.

RT-PCR analysis to determine the expression profiles of WWP RGAs. Total RNA was purified from foliage tissues and reverse-transcribed. Only positive results obtained with the indicated gene-specific primer pairs are shown
Discussion
In the first study aimed at elucidating the diversity and evolution of RGAs in a conifer genome, we report here the characterization of NBS sequences from western white pine. In conifers, RGAs encode TIR-NBS-LRR proteins with the structural features that are also characteristic of angiosperm R proteins. This is consistent with previous studies (Cannon et al. 2002, Meyers et al. 2002), in which RGA searches in GenBank revealed the presence of NBS-LRR sequences in other gymnosperm species. The identification of an NBS-LRR superfamily in both conifers and angiosperms suggests that its common ancestor was present prior to the split between these seed plants about 275 to 300 million years ago (Cannon et al. 2002, Meyers et al. 2002).
Our results indicate that the PCR approach is also useful for systematically searching for RGAs in a conifer genome. Despite the fact that all of the cloned RGAs belong to the TIR-NBS-LRR subfamily, there is great diversity among them at both the nucleotide and amino acid sequence levels (Fig. 1 and Fig. 2). PCR with more primers based on different conserved motifs in the NBS domain should allow us to recover all NBS sequences from the WWP genome. A classical genetic study has demonstrated that at least one R-locus (Cr2) is present in the WWP genome (Kinloch et al. 1999). Although many RGAs might have no functional activity due to disruption of the reading frame, the WWP RGAs cloned in this study resemble angiosperm R genes and encode proteins with the expected structure (Fig. 1 and Table 3). Therefore, it is reasonable to speculate that some WWP RGAs described here may act as functional R genes against pathogens.
The current study finds that almost half of the cloned RGAs are pseudogenes, and some of these are even transcribed into mRNA (Table 3). Sequence comparisons suggested that the WWP RGA pseudogenes originated from point mutations, insertions, or deletion of nucleotides. In a similar study in tomato, only 10% of cloned RGAs turned out to be pseudogenes (Pan et al. 2000b)—a much lower fraction than reported even for other angiosperm species. This may reflect a difference in RGA organization between genomes of angiosperms and conifers. Pseudogenes are nearly as abundant as functional genes in the human genome (Harrison et al. 2002). They have also been identified in the paralogs of the R genes Xa21, Cf9 , Pto and Dm3. Probably serving as reservoirs of potential variation, pseudogenes are believed to be advantageous because they may allow for recombination and gene conversion between alleles or paralogs of functional R genes (Michelmore and Meyers 1998). A recent report has demonstrated that a mouse pseudogene is expressed and regulates the stability of the mRNA expressed from the corresponding protein-coding gene (Hirotsune et al. 2003). Many WWP RGA pseudogenes were found to be expressed in foilage tissues; therefore, investigation of their biological roles may help elucidate the mechanisms of R gene regulation.
In many angiosperm species, RGAs are found clustered around known R loci. Within a cluster of RGAs, there may be more than one gene conferring resistance to different isolates of a specific pathogen or to biologically diverse pathogen taxa (Cooley et al. 2000, Van der Vossen et al. 2000). Some RGAs are genetically linked to R loci in Arabidopsis (Aarts et al. 1998). Therefore, RGA cloning and identification in the WWP genome offers an excellent starting point for a map-based approach to the cloning of target RGA genes, as well as other members in these clusters, including potential R genes. Considering their high level of polymorphism and diversity, and the putatively functional character of their transcription activity, these WWP RGAs will provide a source for the development of molecular markers, especially for the selection of resistance to white pine blister rust (C. ribicola) in forest breeding. For a more definitive elucidation of the proximity of RGA sequences to known R genes in the WWP genome, we intend to construct a genetic map of RGAs, Cr2 and other R genes, merged with general RAPD and AFLP markers.
The proteins encoded by R genes are thought to recognize specific Avr proteins produced by pathogens and activate signal transduction cascades for complex defense responses (Innes 1998, Grant et al. 2000). In these signaling processes, NBS domains play an important role. Diversity among NBS sequences is believed to be critical for the specificity of interaction either with a pathogen elicitor or with proteins downstream in the various signaling pathways that have been identified by genetic analysis (Aarts et al. 1998). Other studies using precise domain swapping have shown a number of LRRs to be essential for the interaction with avirulent factors (Van Der Hoorn et al., 2001; Wulff et al., 2001). Based on NBS sequences, the cloned RGAs in the WWP genome can be grouped into 14 classes (Fig. 2 and Table 3). The high degree of variation in NBS domains suggests that it might contribute in part to the evolutionary fitness of conifers.
R genes are under constant evolutionary pressure because they function to recognize novel Avr gene products produced by the evolving pathogen. Elucidation of the evolution of R genes is very important if we are to uncover the mechanisms by which plants maintain and adapt their defense to pathogens (Pan et al. 2000a). The evolution of R genes remains largely unexplored, but useful information has recently been gained from molecular genetic analyses. Various genetic mechanisms have been proposed to account for the evolution of R genes (Michelmore and Meyers 1998, Noël et al. 1999). It has been hypothesized that point mutations are the major evolutionary mechanism that leads to novel polymorphisms at R-loci (Michelmore and Meyers 1998). Sequence mispairing and unequal crossing-over can also account for R gene evolution (Noël et al. 1999). These analyses refer to crop and modern plants with short life cycles, and so far little data have been obtained from perennial plants like conifers. Our study shows that single nucleotide differences, insertions, deletions and duplications are also found in WWP RGAs, which is consistent with previous observations in angiosperms that revealed that R genes exhibit relatively high rates of mutations and recombination. For long-lived species such as conifers, with a life span of hundreds to thousands of years, the generation of new resistances via somatic mutations may be particularly advantageous (Michelmore and Meyers 1998). Comparisons of variability in the R gene domains that determine pathogen specificity among long-lived individuals need to be performed.
The ratio of nonsynonymous to synonymous changes (Ka/Ks) is an indicator of the evolutionary pressures on a class of genes. Within coding regions, a Ka/Ks ratio greater than 1 reflects diversifying selection and a value lower than 1 suggests purifying selection (Parniske et al. 1997). According to this theory, when under strong selection to generate new variants, R genes should display a pattern in which nonsynonymous substitutions occur more frequently. In the R protein domains, Ka/Ks ratios greater than 1 are generally observed in the β-strand/β-turn motifs of the LRR domain, providing evidence for diversifying selection. This is consistent with the prediction that the amino acids of these LRR motifs might be solvent-exposed and involved in ligand binding for R gene function (Graham et al. 2002). Other regions of resistance genes, like the NBS domains, typically do not show evidence of diversifying selection. Ka/Ks for NBS domains of known angiosperm R genes is about 0.37–0.44, suggesting that the NBS region is subject to purifying selection (Michelmore and Meyers 1998). In contrast to these observations, some Ka/Ks ratios in WWP NBS domains are significantly higher than 1, while others are not significantly different from 1 (Table 4). This evidence for different selection pressures on NBS domains suggests that the evolution of RGAs or R genes might differ between angiosperms and conifers.
References
Aarts MGM, Hekkert BT, Holub EB, Beynon JL, Stiekema WJ, Pereira A (1998) Identification of R-gene homologous DNA fragments genetically linked to disease resistance loci in Arabidopsis thaliana. Mol Plant-Microbe Interact 11:251–258
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Cannon SB, Zhu H, Baumgarten AM, Spangler R, May G, Cook DR, Young ND (2002) Diversity, distribution, and ancient taxonomic relationships within the TIR and non-TIR NBS-LRR resistance gene subfamilies. J Mol Evol 54:548–562
Collins NC, Webb CA, Seah S, Ellis JG, Hulbert SH, Pryor A (1998) The isolation and mapping of disease resistance gene analogs in maize. Mol Plant-Microbe Interact 11:966–978
Creusot F, Macadre C, Ferrier-Cana E, Riou C, Geffroy V, Sevignac M, Dzron M, Langin T (1999) Cloning and molecular characterization of three members of the NBS-LRR subfamily located in the vicinity of the Co-2 locus for anthracnose resistance in Phaseolus vulgaris. Genome 42:254–264
Cooley M, Pathirana S, Wu HJ, Kachroo P, Klessig D (2000) Members of the Arabidopsis HRT/RPP8 family of resistance genes confer resistance to both viral and oomycete pathogens. Plant Cell 12:663–676
Dangl JL, Jones DG (2001) Plant pathogens and integrated defence responses to infection. Nature 411:826–833
Davidson JJ, Ekramoddoullah AKM (1997) Analysis of bark protein in blister rust-resistant and susceptible western white pine (Pinus monticola). Tree Physiol 17:663–669
Ellis J, Jones D (1998) Structure and function of proteins controlling strain-specific pathogen resistance in plants. Curr Opin Plant Biol 1:288–293
Ekramoddoullah AKM, Hunt RS (1993) Changes in protein profile of susceptible and resistant sugar pine foliage infected with the white pine blister rust fungus Cronartium ribicola. Can J Plant Pathol 15:259–264
Ekramoddoullah AKM, Hunt RS (2002) Challenges and opportunities in studies of host-pathogen interactions in forest tree species. Can J Plant Pathol 24:408–415
Ekramoddoullah AKM, Tan Y (1998) Differential accumulation of proteins in resistant and susceptible sugar pine (Pinus lambertiana) seedlings inoculated with white pine blister rust fungus (Cronartium ribicola). Can J Plant Pathol 20:308–318
Ekramoddoullah AKM, Davidson JJ, Taylor D (1998) A protein associated with frost hardiness of western white pine is up-regulated by infection in the white pine blister rust pathosystem. Can J For Res 28:412–417
Ferrier-Cana E, Geffroy V, Macadre C, Creusot F, Imbert-Bollore P, Sevignac M, Langin T (2003) Characterization of expressed NBS-LRR resistance gene candidates from common bean. Theor Appl Genet 106:251–261
Graham MA, Marek LF, Shoemaker RC (2002) PCR sampling of disease resistance-like sequences from a disease resistance gene cluster in soybean. Theor Appl Genet 105:50–57
Grant M, Brown I, Adams S, Knight M, Ainslie A, Mansfield J (2000) The RPM1 plant disease resistance gene facilitates a rapid and sustained increase in cytosolic calcium that is necessary for the oxidative burst and hypersensitive cell death. Plant J 23:441–450
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Harrison PM, Hegyi H, Balasubramanian S, Luscombe NM, Bertone P, Echols N, Johnson T, Gerstein M (2002) Molecular fossils in the human genome: identification and analysis of the pseudogenes in chromosomes 21 and 22. Genome Res 12:272–280
Hirotsune S, Yoshida N, Chen A, Garrett L, Suglyama F, Takahashi S, Yagami K-I, Wynshaw-Boris A, Yoshiki A (2003) An expressed pseudogene regulates the messenger-RNA stability of its homologous coding gene. Nature 423:91–96
Hunt RS (1997) Relative value of slow-canker growth and bark reaction as resistance responses to white pine blister rust. Can J Plant Pathol 19:352–357
Innes RW (1998) Genetic dissection of R gene signal transduction pathways. Curr Opin Plant Biol 1:229–304
Kanazin V, Marek LF, Shoemarker RC (1996) Resistance gene analogs are conserved and clustered in soybean. Proc Natl Acad Sci USA 93:11746–11750
Kinloch BB, Sniezko RA, Barnes GD, Greathouse TE (1999) A major gene for resistance to white pine blister rust in western white pine from the western Cascade Range. Phytopathology 89:861–867.
Kuhner MK, Felsenstein J (1994) A simulation comparison of phylogeny algorithms under equal and unequal evolutionary rates. Mol Biol Evol 11:459–68
Leister D, Ballvora A, Salamini F, Gebhardt C (1996) A PCR-based approach for isolating pathogen resistance genes from potato with potential for wide application in plants. Nature Genet 14:421–429
Leister D, Kurth J, Laurie DA, Yano M, Sasaki T, Devos K, Graner A, Schulze-Lefert P (1998) Rapid reorganization of resistance gene homologues in cereal genomes. Proc Natl Acad Sci USA 95:370–375.
Liu J-J, Ekramoddoullah AKM (2003) Root-specific expression of a western white pine PR10 gene is mediated by different promoter regions in transgenic tobacco. Plant Mol Biol 52:103–120.
Liu J-J, Ekramoddoullah AKM, Yu X (2003) Differential expression of multiple PR10 proteins in western white pine following wounding, fungal infection and cold-hardening. Physiologia Plantarum, in press
Meyers BC, Dickerman AW, Michelmore RW, Sivaramakrishnan S, Sobral BW, Young ND (1999) Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant J 20:317–332
Meyers BC, Morgante M, Michelmore RW (2002) TIR-X and TIR-NBS proteins: two new families related to disease resistance TIR-NBS-LRR proteins encoded in Arabidopsis and other plant genomes. Plant J 32:77–92
Michelmore RW, Meyers BC (1998) Clusters of resistance genes evolve by divergent selection and a “birth-and-death” process. Genome Res 8:1113–1130
Noël L, Moores TL, van der Biezen EA, Parniske M, Dzaniels MJ, Parker JE, Jones JDG (1999) Pronounced intraspecific halpotype divergence at the RPP5 complex disease resistance locus of Arabidopsis. Plant Cell 11:2099–2111
Pan Q, Wendel J, Fluhr R (2000a) Divergent evolution of plant NBS-LRR resistance gene homologues in dicot and cereal genomes. J Mol Evol 50:203–213
Pan Q, Liu Y-S, Budai-Hadrian O, Sela M, Carmel-Goren L, Zamire D, Fluhr R (2000b) Comparative genetics of nucleotide binding site-leucine rich repeat resistance gene homologues in the genomes of two dicotyledons: tomato and Arabidopsis. Genetics 155:309–322
Parniske M, Hammond-Kosack KE, Golsten C, Thomas CM, Jones DA, Harrison K, Wulff BBH, Jones IDG (1997) Novel disease resistance specificities result from sequence exchange between tandemly repeated genes at the Cf-4/9 locus of tomato. Cell 91:821–832
Peñuela S, Danesh D, Young ND (2002) Targeted isolation, sequence analysis, and physical mapping of non TIR NBS-LRR genes in soybean. Theor Appl Genet 104:261–272
Richly E, Kurth J, Lerster D (2002) Mode of amplification and reorganization of resistance genes during recent Arabdopsis thaliana evolution. Mol Biol Evol 19:76–84
Rivkin MI, Vallejos CE, McClean PE (1999) Disease-resistance related sequences in common bean. Genome 42:41–47
Robinson R, Sturrock RR, Davidson JJ, Ekramoddoullah AKM, Morrison D (2000) Detection of endochitinase-like protein in roots of Douglas-fir infected with Armillaria ostoyae and Phellinus weirii. Tree Physiol 20:493–502
Sambrook J, Fritsch E F, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Shen KA, Meyers BC, Islam-Faridi MN, Chin DB, Stelley DM, Michelmore RW (1998) Resistance gene candidates identified by PCR with degenerate oligonucleotide primers map to clusters of resistance genes in lettuce. Mol Plant-Microbe Interact 11:815–823
Speulman E, Bouchez D, Holub EB, Beynon JL (1998) Disease resistance-gene homologs correlate with disease resistance loci of Arabidopsis thaliana. Plant J 14:467–474
Van der Hoorn RA, Roth R, Joosten MHAJ (2001) Identification of distinct specificity determinants in resistance protein Cf-4 allows construction of a Cf-9 mutant that confers recognition of avirulence protein AVR4. Plant Cell 13:273–285
Van der Vossen EA, van der Voort JN, Kanyuka K, Bendahmane A, Sandbrink H, Baulcombe DC, Bakker J, Stiekema WJ, Klein-Lankhorst RM (2000) Homologues of a single resistance-gene cluster in potato confer resistance to distinct pathogens: a virus and a nematode. Plant J 23:567–576
Wulff BB, Thomas CM, Smoker M, Grant M, Jones JD (2001) Domain swapping and gene shuffling identify sequences required for inducting of an Avr-dependent hypersensitive response by the tomato Cf-4 and Cf-9 proteins. Plant Cell 13:255–272.
Young ND (2000) The genetic architecture of resistance. Curr Opin Plant Biol 3:285–290.
Yu YG, Buss GR, Maroof MA (1996) Isolation of a superfamily of candidate disease resistance genes in soybean based on a conserved nucleotide-binding site. Proc Natl Acad Sci USA 93:11751–11756.
Acknowledgements
This research was funded by the Canadian Forestry Service, Canadian Biotechnology Strategy and Forest Innovation Investment awarded to A.K.M.E. We thank Miss Summer Lane for her technical assistance on plasmid extraction and restriction analysis (supported by the Government of Canada Youth Internship Program) and two anonymous reviewers for thoughtful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Hagemann
Rights and permissions
About this article
Cite this article
Liu, JJ., Ekramoddoullah, A.K.M. Isolation, genetic variation and expression of TIR-NBS-LRR resistance gene analogs from western white pine (Pinus monticola Dougl. ex. D. Don.). Mol Genet Genomics 270, 432–441 (2004). https://doi.org/10.1007/s00438-003-0940-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-003-0940-1