
Abstract
Leishmaniasis is responsible for approximately 65,000 annual deaths. Despite the mortality data, drugs available for the treatment of patients are insufficient and have moderate therapeutic efficacy in addition to serious adverse effects, which makes the development of new drugs urgent. To achieve this goal, the integration of kinetic and DSF assays against parasitic validated targets, along with phenotypic assays, can help the identification and optimization of bioactive compounds. Pteridine reductase 1 (PTR1), a validated target in Leishmania sp., is responsible for the reduction of folate and biopterin to tetrahydrofolate and tetrahydrobiopterin, respectively, both of which are essential for cell growth. In addition to the in vitro evaluation of 16 thiazolidine-2,4-dione derivatives against Leishmania major PTR1 (LmPTR1), using the differential scanning fluorimetry (ThermoFluor®), phenotypic assays were employed to evaluate the compound effect over Leishmania braziliensis (MHOM/BR/75/M2903) and Leishmania infantum (MHOM/BR/74/PP75) promastigotes viability. The ThermoFluor® results show that thiazolidine-2,4-dione derivatives have micromolar affinity to the target and equivalent activity on Leishmania cells. 2b is the most potent compound against L. infantum (EC50 = 23.45 ± 4.54 μM), whereas 2a is the most potent against L. braziliensis (EC50 = 44.16 ± 5.77 μM). This result suggests that lipophilic substituents on either—meta and/or—para positions of the benzylidene ring increase the potency against L. infantum. On the other hand, compound 2c (CE50 = 49.22 ± 7.71 μM) presented the highest selectivity index.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Leishmaniasis is an infectious disease caused by protozoa of the genus Leishmania sp., which is endemic in more than 98 countries, distributed throughout Africa, Asia, and Latin America (WHO 2017; 2019). According to World Health Organization (WHO 2019), the incidence of new cases of leishmaniasis per year is estimated at 700,000 to 1 million, and up to 65 thousand deaths are caused by the different clinical forms (WHO 2019).
Despite the public health impact, the therapeutic arsenal to fight leishmaniasis is limited, what underscores the need for new alternatives to treat the patients infected with different species of Leishmania (Gontijo and Carvalho 2003; Swinney and Anthony 2011; Alvar and Arana 2018). One way to achieve this goal is through the inhibition of targets that are unique for the parasite (Eggert 2013). Leishmania genus is auxotrophic for free or conjugated pteridines, such as folic acid and biopterin, which are cofactors in the process of DNA synthesis only in their fully reduced forms (tetrahydrofolate and tetrahydrobiopterin, respectively) (Nare et al. 2009; Leprohon et al. 2015). As a consequence, these parasites developed a complex and versatile biochemical pathway capable of using conjugated and unconjugated pteridines from the host (Nare et al. 1997; Gourley et al. 1999).
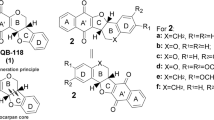
In Leishmania sp., the preferred route to obtain tetrahydrofolate (H4F) is through the action of the enzyme dihydrofolate reductase-thymidylate synthase (DHFR-TS; E. C 1.5.1.3/2.1.1.45), which is considered a validated target for the discovery of new drugs. However, the use of classic antifolates such as methotrexate and trimethoprim is ineffective for the treatment of patients with leishmaniasis. The therapeutic failure is associated with the presence of pteridine reductase 1 (PTR1; EC 1.5.1.33) (Nare et al. 1997). Gene-silencing assays showed that PTR1 is essential for the growth and development of the parasite (Cavazzuti et al. 2008). The selective inhibition of PTR1 is sufficient to kill the parasite, what suggests this is a promising target for the development of new antileishmanial drugs (Vickers and Beverley 2011). Leite et al. (2016) showed that thiazolidine-2,4-dione derivatives are micromolar inhibitors of Leishmania major PTR1 that display competitive behavior with the enzyme cofactor (NADPH) (Fig. 1). In order to improve the compound’s biological profile, 16 novel thiazolidine-2,4-dione derivatives were designed, synthesized, and evaluated against L. major PTR1. This most active compound (4 Kd = 18.56 μM) has a 1.3-fold-higher affinity for LmPTR1 than our previous hit (2d Kd = 25.35 μM). In addition, cell-based assays suggest that high/moderate selectivity (Leishmania braziliensis (MHOM/BR/75/M2903) and Leishmania infantum (MHOM/BR/74/PP75) versus WI-26VA4 fibroblast cells) can be achieved, depending on the substituents decorating the benzylidene ring.

Inhibition profile of 2d against LmPTR1. Kinetic experiments were conducted in the presence of increasing concentrations of the inhibitor: ● = 0 μM; Δ = 25 μM; ☐ = 45 μM. a [Biopterin] = 40 μM. b [NADPH] = 40 μM
Materials and methods
Chemistry
Experiment
All melting points were measured in a capillary tube on a Buchi apparatus. Infrared spectra (1% KBr, cm−1) pellets were recorded on a Bruker IFS66 spectrophotometer and are uncorrected. The 1H NMR and 13C NMR spectra were recorded on a VARIAN VNMRS 400-MR, using 400 MHz for 1H and 75.4 MHz in DMSO-dδ, using tetramethylsilane (TMS) as the internal standard. Chemical shift values are reported in parts per million units. The 13C NMR in CDCl3 and DMSO was maintained at 25 °C using Me4Si (TMS) as an internal standard. The following abbreviations were used to indicate the peak multiplicity: s (singlet), d (doublet), dd (double doublet), ddd (double doublet, doublet), t (triplet), and m (multiplet). The chemical shifts were reported in δ units, and the coupling constants (J) were reported in Hertz. Mass spectra were recorded on a Varian MAT 711 spectrometer 70-eV electron impact. Thin-layer chromatography (TLC) was performed on pre-coated silica plates (Merck Kiesegel 60 F254), and the spots were visualized under ultravioleta light (254 nm/long (365 nm) UV wavelength). The chemical reagents were supplied by Sigma-Aldrich (USA) and were used without further purification. Purity of the compounds was confirmed by using TLC (Kiesel gel 60 G F 254) using the appropriated system for each compound.
Procedure for preparation synthesis of thiazolidine-2,4-dione
The thiazolidine-2,4-dione was obtained by using the method described by Libermann in 1948 and modified by Albuquerque in 1995 (Albuquerque et al. 1995; Libermann et al. 1948; Gouveia et al. 2009). This reaction occurs by condensation of monochloroacetic acid and thiourea in an aqueous medium under reflux for 24 h (Bozdag-Dündar et al. 2007; Bruno et al. 2002) (Fig. 2). Molecular formula C3H3O2NS; yield 78%; mp 118–120 °C; Rf 0.48 (0.9:0.1 CHCl3/MeOH). Recrystallization: water.

Synthetic route of thiazolidine-2,4-dione compounds
General method for the synthesis of 5-arylidene-thiazolidine-2,4-diones (1–16)
The compounds of 1–16, in turn, have the description of the synthesis below.
General method for the synthesis of thiazolidine-2,4-dione
The compounds (1–16) were synthesized in two steps. In step 1, the thiazolidine 2,4-dione compound was synthesized using monochloric acetic acid and thiourea. The solvent used was water. The reagents were heated at 100 °C for 16 h. The compounds 2a-s was prepared by using a simple method, which involve the Knoevenagel condensation with different aromatic aldehydes. The structures of the desired compounds were determined by 1H NMR, 13C NMR, IR, and mass spectrometry.
General method for the synthesis of 5-arylidene-thiazolidine-2,4-dione (1–16)
The compounds 1–16 and compounds 2a–2e were synthesized from a mixture of thiazolidine-2,4-dione (1—Fig. 2) (2.5 g, 21.36 mmol), aldehyde (21.36 mmol), piperidine (14.11 mmol), and ethanol (150 mL). The reaction mixture was heated under reflux and continuously stirred for a period of 16 h. The course of the reaction was monitored by using TLC. The reaction mixture was poured into water and acidified with acetic acid. The resulting precipitate was filtered off and recrystallized from acetic acid to give (2—Fig. 2).
(Z)-5-(3-methoxy-4-hydroxy-benzilydene)-thiazolidine-2,4-dione (1)
Yield 70%; mp 260 °C; Rf 0.55(CH2Cl2/MeOH 9:1) Recrystallization: ethanol; IR (KBr 1%, νmax cm−1) 1566 (C=C); 1730–1670 (C=O). 1H NMR (CDCl3, 300 MHz, δ ppm): 8.48 (s 1H, NH); 7.71 (s 1H, CH=); 3.77 (s 3H, OCH3); 6.02 (s 1H, OH). 6.92 (s 1H(2)); 6.86 (d 1H(5) J = 8.33); 6.91 (d 1H(6) J = 8.34); 13C NMR (Acetone-d6, 75.4 MHz, δ ppm): 169.81 (C=O2); 166.89 (C=O4); 119.55 (C5, heterocycle); 143.28 (CH=); 56.50 (OCH3); 129.20 (C1); 112.58 (C2), 148.06 (C3); 144.76 (C4); 115.56 (C5); 122.30 (C6). Anal. Calcd. for C11H9NO4S: C, (52.58%); H, (3.61%); N, (5.57%); Found: C, 53.01%; H, 3.83%; N, 5.25%. HRMS+: calcd 251.0252; found 251.0253.
(Z) 5-(2,4-dimethoxy-arylidene)-thiazolidine-2,4-dione (2)
Yield 67%; MP 250 °C; MF C12H11NO4S; MW 265.2850; Rf 0.50 (CHCl3/MeOH 9:1 Recrystallization: ethanol; IR (KBr 1%, νmax cm−1) 1571 (C=C); 1733–1691 (C=O). 1H NMR (CDCl3, 300 MHz, δ ppm): 11.30 (s 1H, NH); 8.25 (s 1H, CH=); 3.86 (s 3H, OCH3 (orto); 6.57 (s 1H, H3); 3.87 (s 3H, OCH3 para); 6.0 (d 1H(6) J = 8.34); 11.40 (s 1H, NH, (imide). HRMS+, calcd 250.0048; found 265.0409.
(Z) 5-(2-nitro-benzylidene)-thiazolidine-2,4-dione (3)
Yield 83%; MP 205–206 °C; MF C10H6N2O4S; MW 250.2306; Rf 0.50 (Hex: Ethyl acetate 9:1) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1) 1569 (C=C); 1736–1660 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 11.97 (s 1H, NH (imide)); 8. 57 (d 1H C-H), 7.81 (dd 2H, H4, J = 7.3), 7.77 (dd 1H, H5) J = 7.6; J = 1.5, 8.13 d,1H J = 7.6 (H6). HRMS+, calcd. 250.0048; found 250.0049.
(Z) 5-(2,3,5-Trichloro-benzylidene)-thiazolidine-2,4-dione (4)
Yield 73%; MP 170.5–171.0 °C; MF C10H4Cl3NO2S; MW 308.5683. Rf 0.50 (CHCl3/MeOH 9:1) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1) 1569 (C=C); 1738–1667 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 11.97 (s 1H, NH (imide)); 8. 35 (s 1H C=H), 7.55 (s 1H, (H4). 7.23 (s 1H, H6) HRMS+, calcd. 306.9028; found 306.9028.
(Z)-5-(3-methoxybenzylidene) thiazolidine-2,4-dione (5)
Yield 83%; MP 170–171 °C; MF C11H9NO3S; MW 235.2591; Rf 0.50 ((Hex: Ethyl acetate 5:5) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1) 1564 (C=C); 1733–1665 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 11.87 (s 1H, NH (imide)); 7.87 (s 1H C=H), 7.19 (d 1H, H2–4, J = 7.5), 3.83 (s 3H OCH3) 7.56 (t 1H, H5) J = 7.6; J = 1.5), 6.90.(d,1H J = 7.6 (H6). HRMS+, calcd. 235.0303; found 235.0303.
(Z) 5-(3-hydroxy-benzylidene)-thiazolidine-2,4-dione (6)
Yield 69%; MP 265–266 °C; MF C10H7NO3S; MW 221.2025; Rf 0.50 ((Hex: Ethyl acetate 5:5) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1) 1568 (C=C); 1738–1669 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 12.31 (s 1H, NH (imide)); 7.97 (s 1H C=H), 6.72 (d 1H, H2, J = 7.48), 5.37 (s 1H OH) 6.86 (d 1H, H4) 7.56.(s 1 H) 7.56; J = 1.5), 7.19 (d,1H, J = 7.16; (H6). (d,1H) HRMS+, calcd. 235.0303; found 235.0303.
(Z) 5-(3,4-dimethoxy-benzylidene)-thiazolidine-2,4-dione (7)
Yield 73%; MP 213–214 °C; MF C12H11NO4S; MW 265. 0409; Rf 0.46 ((Hex: Ethyl acetate 7:3) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1) 1568 (C=C); 1735–1668 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 12.31 (s 1H, NH (imide)); 7.95 (s 1H C=H), 7.24 (s 1H, H2), 3.84 (s 3H OCH3) 3.86 (s 3H, OCH3) 6.96 (d,1H, J = 7.7; (H5). 7.21 (d,1H, J = 7.6; (H6). HRMS+, calcd. 265.0409; found 265.0409.
(Z) 5-(4-methylthio)-benzylidene)-thiazolidine-2,4-dione (8)
Yield 63%; MP 209–210 °C; MF C11H9NO2S2; MW 251.3247–251.3251. Rf 0.65 (Hex: Ethyl acetate 6:4) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1) 1571 (C=C); 1735–1668 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 12.31 (s 1H, NH (imide)); 7.95 (s 1H C=H), 7.24 (s 1H, H2), 3.84 (s 3H OCH3) 3.86 (s 3H, OCH3) 6.96 (d,1H, J = 7.7; (H5). 7.21 (d,1H, J = 7.6; (H6). HRMS+, calcd. 265.0409; found 265.0409.
(Z) 5-(2-Chloro benzylidene)-thiazolidine-2,4-dione (9)
Yield 70%; MP 165.9–166.5 °C; MF C10H6ClNO2S; MW 239.6781. Rf 0.50 (Hex: Ethyl acetate 6:4) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1) 1568 (C=C); 1733–1667 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 12.29 (s 1H, NH (imide)); 8.23 (s 1H C=H); 7.54 (d 1H, H3 J = 7.5; 1.4), 7.05 (t 3H (H4) J = 7.4; 1.5) H5 7.25 (t 2H, (H5) J = 7.4; 1.5) 7.28 (d,1H, J = 7.7) (H5). 7.28 (d,1H, J = 7.6; (H6). HRMS+, calcd. 238.9808.; found 238.9808.
(Z) 5-([1,1′-biphenyl]-4-ylmethylene) thiazolidine-2,4-dione (10)
Yield 78%; MP 238.5–239.8 °C; MF C16H11NO2S; MW 281.0510. Rf 0.53 (CH2Cl2/Acetone (0.53) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1) 1567 (C=C); 1731–1665 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 12.29 (s 1H, NH (imide); 7.99 (s 1H C=H); 7.56 (d 1H, H2-H2’;J = 7.5; 1.4), 7.74. (d 2H (H3-H3’) J = 7.4; 1.5) 7.40 (t 2H (H6-H6’) J = 7.5–1.50; 1.50. (t 2H, (H7−H7’) J = 7.6; 1.5; 1.5). 7.40 (t 2H, (H6−H6’) J = 7.6; 1.5; 1.5). HRMS+, calcd. 281.0510.; found 281.0510.
(Z) 5-((1H-indol-3-yl) methylene) thiazolidine-2,4-dione (11)
Yield 78%; MP 165.9–166.5 °C; MF C12H8N2O2S; MW 239.6781. Rf 0.50 (Hex: Ethyl acetate 6:4) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1) 1567 (C=C); 1733–1667 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 12.29 (s 1H, NH (imide)); 8.11 (s 1H C=H); 7.76 (d 1H, H2 indol J = 7.5; 1.4); 12.0 (d 1H (NH)) 7.05 (d 1H (H1, Ar) J = 7.4; 1.5); 7.11 (t 2H, (H2) J = 7.4; 1.5; 1.5) 7.18 (t, 2H, (H3). J = 7.7; 1.5, 1.5)) 7.57 (d, 1H, J = 7.6, H6). HRMS+, calcd. 244.0306; found 244.0306.
(Z)5-((6-methylene-4-oxo-4H-chromen-3-yl) methylene)) thiazolidine-2,4-dione (12)
Yield 72%; MP 287.5- 0C; MF C14H9NO4S; MW 287.2906. Rf 0.5 ((Hex: Ethyl acetate 6:4) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1) 1559 (C=C); 1730–1666 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 12.29 (s 1H, NH (imide)); 7.43 (s 1H C=H); 6.71 (s 1H (H1)) 7.63 (d 1H (H2)) 7.67 (d 1H (H3), Ar) J = 7.47.2.44 (s 3H, (CH3) 7.41 (s 1H), HRMS+, calcd. 289.0409; found 289. 0409.
(Z)-5-((4-nitro-1H-indol-3-yl)methylene)thiazolidine-2,4-dione (13)
Yield 57%; MP 129.5–131.0 0C; MF C12H7N3O4S; MW 289.2667; Rf 0.54 (Dichloro metane: Methanol 9.9:0.1) Recrystallization: ethanol, IR (KBr 1%, νmax cm-1) 1560 (C=C); 1729–1657 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 12.29 (s 1H, NH (imide)); 7.21 (s 1H C-H); 7.95 (t 2H (H2) J = 7.63, J = 1.4 J = 1.6). 7.23 (d 1H (H3) J = 7.47); 9.9 (d 1H J = 7.80 (NH), 8.81 (d 1H, J = 7.79). HRMS+, calcd. 289.0157; found 289.0156.
(Z)-5-(quinolin-4-ylmethylene)-thiazolidine-2,4-dione (14)
Yield 83%; MP 273–274 °C; MF C14H10N2O2S; MW 270.3036. Rf 0.50 ((Hexane Ethyl acetate 9:1) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1) 1559 (C=C); 1733–1661 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 12.26 (s 1H, NH (imide)); 7.82 (s 1H C-H); 6.73 (t 2H (H2) J = 7.60, J = 1.5 J = 1.5). 7.76 (t 1H (H3) J = 7.45;1.5,1.5) 8.12 (d 1H (H4), 8.75 (d 1H, J = 7.47, J = 1.5) 7.12 (d 1H, J = 7.3, J = 1.5 HRMS+, calcd. 256.0306; found 256.0306.
(Z)-5-((5-Bromothiophen-2-ylmethylene)-thiazolidine-2,4-dione (15)
Yield 83%; MP 180.3–180.7 °C; MF C8H4BrNO2S2; MW 290.1490. Rf 0.50 (CHCl3/MeOH 9:1) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1) 1558 (C=C); 1735–1662 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 12.25 (s 1H, NH (imide)); 7.55 (s 1H C-H); 7.55 (d 1H (H1, C=H J = 7.50); 7.24 (d 2H (H2,-H2´ J = 7.46; J = 1.45; J = 1.45) 7.45 (d d H3-H5 CH) J = 7.24. HRMS+, calcd. 288.01878; found 288.01878.
(Z)-5-((E)-2-methyl-phenylallylidene (thiazolidine-2,4-dione) (16)
Yield 79%; MP 204.6–205.2 °C; MF C13H11NO2S; MW 245–246. Rf 0.49 ((Hexane: Ethyl acetate 9.9:0.1) Recrystallization: ethanol, IR (KBr 1%, νmax cm−1); 1555 (C=C); 1733–1665 (C=O). 1HNMR (CDCl3, 300 MHz, δ ppm): 12.27 (s 1H, NH (imide)); 7.41 (C=C; 6.37 (s 1H C-H); 2.21 (s 3H CH3); 6.34 (s 1H CH) 7.61 (d 2H (H2,-H2`) J = 7.46; J = 1.45; J = 1.45) 7.45 (d 2H (H3-H5´) CH) J = 1.45; J = 1.45) 7.33 (t 2H. HRMS+, calcd. 245.0510 found 245.0510.
The 5-arylidene thiazolidine-2,4-diones compounds (2a–e) were synthesized as described in Leite et al. 2016.
In vitro assays
ThermoFluor® assays
Thermal shift assays were carried out using a CFX Touch RT-PCR Detection System (Bio-Rad CFX Manager®, Bio-Rad Laboratories, Inc.) fitted with custom filter sets. The data was recorded in the CFX Manager™ v 3.1 Software. Initially, conditions of the assay (choice of buffer, protein concentration, and DMSO concentration) were optimized for LmPTR1. Each parameter was tested in triplicate on a 96-well PCR plate (PCR plates 96-well Bio-Rad®), manually sealed with transparent capping strips (Flatcap strips Bio-Rad®). The final protein concentration, for optimization trials, was set to 5 μM, except for the determination of optimum PTR1 concentration, which varied from 1 to 5 μM. Fluorescence changes were monitored with SYPRO Orange® dye, using 492- and 610-nm wavelengths for excitation and emission respectively. Thermal shift assays were carried out from 25 to 85 °C in increments of 1 °C per minute. Fluorescence data (raw data) was recorded using the CFX Manager™ v 3.1 Software, and then exported to Excel 2007 worksheet (ftp://ftp.sgc.ox.ac.uk/pub/biophysics) for processing and analysis. Tm values employed for compound comparison were those calculated from the non-linear fitted melting curves (Boltzmann sigmoidal function) implemented in GraphPad Prism version 5.0 for Windows (GraphPad® Software, San Diego, CA, USA, www.graphpad.com).
Single-concentration assays
The PTR1 expression and purification steps were carried out as described previously (Leite et al. 2016). All compounds were assayed at the final concentration of 50 μM. In each well, a solution containing 4 μL of LmPTR1 (to obtain the final concentration, 5 μM), 14 μL of 20-mM sodium acetate buffer pH 4.7, 1-μL SYPRO Orange (Invitrogen™) (5X), and 1-μL of each compound was added. The values of Tm obtained in the presence of each compound were compared with the value of the reference solution, which contains 5% DMSO P.A. (v/v), instead of the inhibitor. The assays were performed in triplicate.
Concentration-response assays
Compounds with ΔTm > 1.0 °C, in single-concentration assays, also had their effect evaluated at the following concentrations: 6.25, 12.5, 25, 50, and 100 μM. The obtained Tm values were compared with the value of the reference solution, which contains 5% DMSO P.A. (v/v), instead of the inhibitor. All assays were carried out in triplicate.
Determination of the dissociation constant by ThermoFluor®
Compounds that show a concentration-response behavior, in the previous step, had their dissociation constants (Kd) determined according to the equation established by Vivoli et al. (2014), using data from ThermoFluor assays carried out in the presence of the following concentrations of each compound: 6.25, 12.5, 25, 50, 100.0, 200.0, and 400.0 μM. The five compounds that presented the best ΔTm values in the study by Leite et al. (2016) also had their dissociation constants determined, following the same concentrations and previous parameters of ThermoFluor® assays. The assays were performed in triplicate.
Cellular assays
Cultivation of cell lines
The human cell line WI-26VA4 (pulmonary fibroblast ATCC CCL-75) was used for toxicity analysis. This lineage is part of the animal cell bank of the Cell Biology Service (SBC) of the Ezequiel Dias Foundation (FUNED) in Belo Horizonte - Minas Gerais.
Cells were cultured from cryopreserved ampoule; they were thawed at 37 °C; their contents were transferred to a 15-mL tube containing 10-mL RPMI 1640 medium. Cells were centrifuged at 1200×g for 5 min; the supernatant was discarded, and the pellet was resuspended in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (SBF) (complete medium). The cells were transferred to plastic bottles of treated cell culture to promote cell adhesion, T75 (75 cm2) 10 mL (Corning Costar Inc., USA), and maintained as monolayers at 37 °C in a greenhouse (Thermo electron co.) humid atmosphere of 5% CO2. The culture medium was replaced every 48 h of incubation. Cellular morphology and monolayer formation were observed with the aid of an inverted microscope at × 100 magnification (Olympus model, CKX 41). After confluence of 80% of the culture in the T75 bottle, the cells were replicated or used in the cytotoxicity assays (Patra et al. 2011; Pereira et al. 2012).
Cytotoxicity assays
Cells with 80% confluency were trypsinized with 1.5 mL of trypsin (1:250 Sigma), incubated at 37 °C for 5 min, resuspended in 10 mL of complete RPMI medium, and centrifuged at 1200×g for 5 min. The supernatant was discarded, and the pellet was resuspended with complete RPMI medium. Cells were distributed into 96-well microplates at a density of 4 × 105 cells/100 μL per well and incubated in a CO2 oven at 37 °C for 24 h for adhesion to the plate. After this period of adhesion, the medium was removed, a new RPMI medium supplemented with 1% FBS was added, and then the diluted compounds were added in RPMI medium containing different concentrations (0.01, 0.10, 1.0, 10, and 100 μg/mL). The plates were incubated for an additional 48 h.
At the end of the treatment period, 100 μL/well of a solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazole (MTT) (Sigma-Aldrich) bromide, tetrazole salt, concentration of 0.5 mg/mL in RPMI 1640 without phenol red (Denizot and Lang 1986) were add in the plate. After 3 h of incubation in the CO2 incubator at 37 °C in the dark with MTT, the supernatant was aspirated and the formazan crystals were dissolved in 50 μL/well of DMSO P.A. and held at 37 °C for 10 min. The absorbance per well was measured at a wavelength of 550 nm using the Gen5 software (Data-Analysis Software-Bio-Tek). The assays were performed in triplicate.
Cultivation of Leishmania sp.
The parasite cultures were carried out with reference strains of Leishmania (V.) braziliensis (MHOM/BR/75/M2903) and Leishmania (L.) infantum (MHOM/BR/74/PP75) belonging to the Laboratory of Parasitic Infectious Diseases of the UFSJ/CCO (LAB-DIP/CCO).
The parasites were cultured in biphasic medium, the liquid phase being composed of Schneider’s Insect Medium with L-glutamine and sodium bicarbonate (Sigma®), plus 10% Fetal Bovine Serum (FBS) and 1% antibiotic Pen Strep-GIBCO (10,000 units/mL penicillin and 10,000-μg/mL streptomycin), and maintained in solid phase with NNN culture medium (Novy, McNel, and Nicolle) plus 1% antibiotic Pen Strep for storage. The parasites were kept in bottles for cell culture in polystyrene, growth volume of 25 cm3, sterile, and in BOD greenhouse at 25 °C. For the maintenance of the parasites, peaks were performed every 4 days. All procedures involving the maintenance and cultivation of the strains were carried out in a laminar flow hood, avoiding any type of contamination by microorganisms. Follow-up of the cultures was carried out by observation in a ZEISS optical microscope, AXIOSTAR PLUS model, by the preparation of crop slides, in order to observe flagellar mobility and absence of contamination.
Antileishmania activity assays
Antileishmania activity assays were performed by the same cytotoxicity test, i.e., the MTT chemosensitivity test. After 48 h of boiling, to allow the parasites to grow, that is, in the log phase (ideal phase for testing the compounds), the viability of the parasites was evaluated by microscopy. Aliquots of the viable parasites were removed from the growth bottles, and the ideal volume was calculated for a final concentration of 1.0 × 106 parasites/well. The reaction volume per well was 200 μL, with 20 μL corresponding to the compounds to be tested (at the concentrations of 1.5625, 3.123, 6.25, 12.5, 25, 50, and 100 μg/mL in triplicate for the compounds and 50 μg/mL for amphotericin B deoxycholate) and 180 μL of parasite at the concentration of 1.11 × 106 parasites/mL for each well in 96-well microplates. From this, the incubation was incubated at 26 °C for 48 h.
At the end of the incubation period, the MTT was diluted in incomplete Schneider’s medium (without fetal bovine serum), so that it was at a concentration of 5 mg/mL. From this moment, the experiments were performed with the laminar flow hood light off, since the MTT is photosensitive. A volume of 20-μL MTT was added to the wells. The plates were then wrapped in aluminum foil and incubated for 4 h at 37 °C. The plates are maintained at this temperature, as the compounds have already acted, the temperature no longer affects the viability of Leishmania protozoan, and some proliferative capacity is diminished. It is also worth noting that in low temperatures, the production of formazan salts is lower.
After 4 h of incubation with MTT, the plates were centrifuged at 3500×g for 10 min at 4 °C in the plate centrifuge. At the end of centrifugation, the supernatant was carefully removed to preserve the formazan crystals and parasites, 50 μL/well of DMSO P.A. was added to dissolve the formazan, and the plates were homogenized and allowed to stand for 15 min for reading in ELISA reader at 570 nm using the Gen5 software (Data-Analysis Software-Bio-Tek). The assays were performed in triplicate.
Calculation of the selectivity index
After determination of the effective concentration to kill 50% of the parasites (CE50) and determination of the lethal dose capable of killing 50% of human cells (LD50), the selectivity index (SI) was calculated according to the equation:
Results and discussion
Synthesis and characterization of thiazolidine-2,4-dione
Leite et al. (2016) showed that thiazolidine-2,4-dione derivatives inhibit LmPTR1 at micromolar range concentration. Compound 2d, the most promising compound identified in that study, was used as a prototype to design novel compounds with optimized effect against Leishmania cells in vitro. A total of 16 compounds have been synthetized based on features of the prototype and their structural variations: compounds 1 to 10 (except for 2, 3, and 6) were synthetized based on chemical viability, while 11–15 were designed to explore additional LmPTR1 pockets nearby the active site. Compounds 2, 3, and 9 were designed to the effect of bulk substituents (steric clashes) on binding affinity, whereas compound 16 aimed at investigating the effect of a spacer moiety, between the two rings on the binding profile.
ThermoFluor® assays
Single-concentration assays for the identification of LmPTR1 inhibitors
Sixteen structural analogs of compound 2d were evaluated for their ability to interact with LmPTR1. ThermoFluor® is a high/medium throughput screening that has enabled the prioritization of potential molecules due to the ability to evaluate the (de)stabilizing effect of hundreds of compounds over the tertiary structure of the protein and, therefore, their ability to bind to the target and generate a biological response. Molecules with ΔTm > 1.0 °C are described as promising and tend to confirm the biological potential in secondary assays (Pantoliano et al. 2001; Ericsson et al. 2006; Cimmperman and Matulis 2011; Kopec and Schneider 2011). This technique has already been employed to identify thiazolidine-2,4-dione that inhibit LmPTR1 (Leite et al. 2016) or that inhibit both DHFR-TS and PTR1 (Teixeira et al. 2019). Compounds with higher degree of affinity to the target tend to have higher ΔTm when all other variables are kept equal (Zaia et al. 1998; Pantoliano et al. 2001; Ericsson et al. 2006; Niesen et al. 2007; Zhang and Monsma 2010). Among the 16 novel thiazolidine-2,4-dione derivatives evaluated by a single-concentration ThermoFluor® assay against LmPTR1 (Graph 1), only two compounds, 4 (ΔTm = 12.12 ± 0.34 °C) and 9 (ΔTm = 4.69 ± 0.21 °C), may be considered as potential inhibitors against pteridine reductase 1 of L. major.

Thermal stability of LmPTR1 in the presence of the thiazolidine-2,4-dione derivatives in the single-concentration assay. The values represent the median and interquartile range of the variation of Tm (ΔTm). Statistical differences were calculated between each compound (n = 3) and compound 2d, ***p < 0.001 (Kruskal-Wallis test with Dunn’s post-test), previously reported by Leite et al. (2016)
However, both compounds have lower ΔTm values than compound 2d, (compound 4 ΔTm = 12.12 ± 0.34 °C and compound 9 ΔTm = 4.69 ± 0.21 °C versus compound 2d ΔTm = 44.67 ± 1.74 μM). Single-concentration assays also show that compound 12 shows negative thermal displacement. Some studies have shown that compounds with this profile can stabilize the target with whom they interact in a conformation other the native one. However, a direct relationship between negative values of thermal shift and biological profile has not been described (Waldron and Murphy 2003; Dupeux et al. 2011; Garbett and Chaires 2012; Sancho 2013; Contessoto et al. 2018).
Thus, only molecules 4 and 9 were evaluated at different concentrations (6.25, 12.5, 25, 50, and 100 μM) to probe if the thermal stabilization follows a dose-dependent behavior. Both compounds were found to increase LmPTR1 thermal stability at higher concentrations, with ΔTm values for compound 4 higher than those seen for compound 6 (Graph 2). For compound 4, a significant increase (p < 0.01) in ΔTm is observed between 6.25-μM and 12.5-μM and between 12.5-μM and 25-μM concentrations. Higher concentrations afford negligible increases in LmPTR1 Tm. Noteworthy is an eight-fold increase in thermal shift between the lowest and highest ligand concentration. In contrast, compound 9 showed a significant increase (p < 0.01) in ΔTm only at low concentrations (between 6.25 μM and 12.5 μM), and despite the overall tendency to increase LmPTR1 TM at higher concentrations, only a 6-fold increase in thermal shift is observed if one compares the lowest and the highest concentration of compound 9.

Effect of different concentrations of compound 4 (a) or 9 (b) over the LmPTR1 thermal stability. The values represent the median and interquartile range of the variation of Tm (ΔTm) by multiple comparisons (n = 3), **p < 0.01. (Kruskal-Wallis test with Dunn post-test)
Determination of apparent dissociation constant by ThermoFluor®
The inhibition constant (Kd) can also be determined by ThermoFluor® assays (Lo et al. 2004). This assay does not require a high degree of sample purity and does not have to include solution enthalpy in the Kd calculation, which sometimes makes it difficult to characterize (Cimmperman and Matulis 2011; Craig and Newton 1991; Vivoli et al. 2014). Alternatively, Vivoli et al. (2014) developed a method to calculate the apparent dissociation constant (Kdapp) from information, provided by differential scanning fluorimetry assays, which in most cases is very close to the real Kd value (Petrauskas et al. 2019).
In order to make a fair comparison of the current compounds to those already published (Leite et al. (2016)), not only compounds 4 and 9 had their Kdapp calculated but also compounds 2a–2e (Table 1).
According to this method, compound 4 (18.56 μM) has the lowest Kdapp value, followed by compound 2b (23.12 μM), 2d (25.35 μM), 2e (89.98 μM), and 9 (98.11 μM). Hence, compound 4 seems to have higher affinity to LmPTR1 than our previous lead compound (2d). Moreover, compounds with Kdapp ≤ 100 μM are expected to have moderate to high affinity for the target (Vivoli et al. 2014). As compound 2d is among the most promising compounds, the assumptions made by Cimmperman and Matulis (2011) and Vivoli et al. (2014) are in good agreement with our previous results (Leite et al. 2016), since the IC50 is related to the ability of a compound to inhibit the catalytic activity of the target at a fixed substrate concentration and Kd accounts for the ability of a ligand to bind with the target.
Cellular assays
It is known that the use of enzymatic assays for the characterization of specific target inhibitors has been commonly used since the advent of molecular cloning (Pikkemaat et al. 2002; Gasteiger et al. 2005; Sievers et al. 2011; Priest and Erdemli 2014). However, this approach is considered a simplistic view of the effect of the compound against the pathology (Grasberger et al. 2005; Lesyk et al. 2006; Vicini et al. 2006; Silva et al. 2014; Wagner and Schreiber 2016). An example to be highlighted is the research carried out by Sienkiewicz et al. (2010), which, after genetic validation of Trypanosoma brucei PTR1 as a potential target, developed a potent and selective inhibitor of TbPTR1 (IC50 for TbPTR1 = 7.0 nM; TbDHFR ≥ 30.0 μM) that is unable to inhibit parasite growth. Thus, enzymatic assays only account for the compound’s ability to interact with the target, whereas its permeability through the cell membranes, efflux process of the compounds, the retention capacity in lipophilic compartments, the high rate non-specific binding to proteins, among other factors remain largely ignored by such assays (Gilbert 2013). Although the in vitro assays were performed on pteridine reductase 1 from Leishmania major, in Brazil, the endemic species responsible for the clinical manifestations of CL, CML, and VL are, mainly, L. braziliensis and L. infantum, respectively. Nevertheless, the NADPH-dependent oxidoreductase enzymes present conservation of their supersecondary structure (Vidal et al. 2018) and highly similar active sites. In fact, a sequential identity analysis of PTR1 (Fig. 3) among the three species reveals 77.8% sequence identity between LmPTR1 and LbPTR1 and 88.9% sequence identity between LmPTR1 and LiPTR1 or LiPTR1 and LbPTR1. Despite overall similarity, the cofactor-binding site has two substitutions, His-241 for a Tyr-241 and a Tyr-194 for a Phe-194, that might impact the binding of ligands. In fact, Tyr-194 is considered an essential amino acid in the mechanism proposed by Gourley et al. (2001). However, previous results from our group (Leite et al. 2016) suggest that this residue does not interact with 2d, which only interact with residues that are conserved in LbPTR1 and LiPTR1. Thus, based on the sequential alignment data as well as the interaction profile described for the 2d, it is reasonable to assume that the L. major PTR1 inhibitors described in this work also bind to PTR1 from other Leishmania species. In order to support this hypothesis, compounds 2a, 2b, 2c, 2d, 2e, 4, and 9 were evaluated against the strains of L. braziliensis and L. infantum. In addition, their cytotoxicity profile in human cells was also evaluated.

Multiple sequential alignment of PTR1 of L. major (LmPTR1), L. infantum (LiPTR1), and L. braziliensis (LbPTR1). Multiple alignment was generated on the online Clustal Omega server. The rectangles delimit the residues present at cofactor binding site. (“*” = Identical, “:” = similar, “.” = not conserved, “-” = absent)
The evaluation of promising compounds against Leishmania sp. consists in determining the efficacy in inhibiting parasite growth by 50% (CE50). However, cytotoxicity assays in human cell lines are essential for evaluating the possible damages caused by the compounds on the human organism and to understand these compound selectivity profile.
Initially the seven compounds were analyzed for cytotoxicity against the WI-26VA4 cell line (ATCC CCL-75 lung fibroblast) in order to determine the dose capable of killing 50% of cells (LD50) (Table 2). The seven compounds evaluated had an LD50 value higher than the drug used as reference (amphotericin B deoxycholate). Compounds 2b and 2c are the least cytotoxic in the series. The high standard deviation of error presented by compound 9 prevents a correct classification of its toxicity. Perhaps the structure of the compound enables it to emit fluorescence at the same wavelength as MTT, causing interference. The action of the compounds on human cells is unknown, since humans do not possess the enzyme PTR1. Although there are NADPH-dependent oxidoreductase enzymes in human cells, this does not guarantee the action of the compounds on them. Teles et al. (2018) have shown that analogous compounds, which bind to DHFR of Schistosoma mansoni (SmDHFR), have a competitive mechanism of inhibition towards the substrate. However, compounds 2a, 2b, and 2c were inactive against the SmDHFR. Thus, it is not possible infer these compounds’ mechanism of action from those studies, since interference with normal physiology can be triggered by the impairment of basic cellular functions, such as energy metabolism, cytoskeleton organization, membrane integrity, glycogen storage, electrical conductivity, glutathione depletion, radical formation, or alteration of uptake or excretion (Schoonen et al. 2005). It should be noted that high standard deviations are characteristic for MTT phenotypic assays (Stockert et al. 2012).
Next, the effective concentration to inhibit 50% growth (EC50) of Leishmania (V.) braziliensis (MHOM/BR/75/M2903) and Leishmania (L.) infantum (MHOM/BR/74/PP75) was determined (Table 2).
For the L. braziliensis species, all compounds show similar EC50 values; however, compound 2c shows higher selectivity for L. braziliensis than the other compounds. In contrast, the biological activity of the compounds against the species of L. infantum exhibited a wider distribution of EC50 values, compound 2b was the most potent (EC50 = 23.5 ± 4.5) and the most selective (SI 1.85). On the other hand, compound 2c is among the least potent ((EC50 = 66.1 ± 14.2), despite its selectivity profile remains high. Thus, compound 2b is the best lead compound. No compound showed superior biological potency than amphotericin B deoxycholate, the control drug for both species.
Given that compound 2b presented the best biological activity (EC50 = 23.45 ± 4.54 μM) and one of the best Kd values (23.12 μM), it seems that the presence of small lipophilic substituents in the meta and/or para position is essential for effective compounds against Leishmania species. Although these compounds have lower biological activity than amphotericin B deoxycholate, they are less cytotoxic. Lipophilic substituents at the orto position affect the affinity of the compounds for the L. infantum species and, consequently, the biological activity. The relationship between the biological affinity of compounds 2a, 2b, 2d, 2e, and 4 against LmPTR1 and the biological potency of these compounds compared with the L. infantum species may be associated with greater conservation of residues at the binding site of LiPTR1 and LmPTR1, since the same behavior is assumed in the mechanism of action of these compounds with the prototype compound 2d. Hence, the conservation of the Tyr-194 residue, essential for the catalytic mechanism of PTR1, in the species LiPTR1 and LmPTR1, may be responsible for the improved activity profile of the compounds against L. infantum (Fig. 4). This line of thought does not hold for the L. braziliensis species, since there is no relationship between structure and activity. This difference, therefore, is expected, since L. major and L. infantum are closer, from the phylogenetic point of view, than L. braziliensis, a neotropical species. Considering the structure-activity relationship data, it can be seen that both Kd and EC50 values follow the same structural relationship profile.

Structure-activity relationship of the 7 compounds tested against L. infantum (EC50). Shaded atoms in blue promote improved potency, and those shaded in red promote potency loss
Following the determination of the LD50 of the compounds against the human cell line and the EC50 against the species of Leishmania, the selectivity index was calculated (Table 2) in order to assess to which organism the compounds are most harmful.
Only compounds 2b for L. infantum and 2c for both L. braziliensis and L. infantum had a selectivity index greater than 1 and were more effective against the parasite than cytotoxic to human cells. Although the literature adopts a value greater than 10 for the selectivity index, in order to consider that a compound is promising in relation to phenotypic assays (Di Giorgio et al. 2004; De Mesquita et al. 2009), this does not mean that compounds with the values below are insignificant. Thus, compound 2b, due to both enzymatic and cellular characteristics, is considered the most promising lead compound.
Conclusions
The knowledge about compounds’ biophysical characteristics is critical to set a suitable screening process. The information provided by LmPTR1 thermal shift assays suggest that compound 2c has low affinity to the target, whereas compounds 2a, 2b, 2d, and 4 have micromolar affinity to LmPTR1. As might be expected, these compounds are also active against other Leishmania species, as they share greater phylogenetic proximity. The most potent compound for the L. infantum is compound 2b (CE50 = 23.45 ± 4.54 μM), while the most potent compound for the L. braziliensis species is compound 2a (CE50 = 44.16 ± 5.77 μM). These results highlight that kinetic characterization is the first step to identify a novel inhibitor; however, subsequent phenotypic assays make it possible to probe the compound behavior in complex media that allow pharmacokinetic parameters to be extrapolated for in vivo assays.
Results from phenotypic assays have been routinely employed, by research groups and the pharmaceutical industry alike, to decide which compound to move forward, or not, based on their pharmacokinetic and toxicological profile. Accordingly, it is found that only compounds 2b and 2c for L. infantum and 2c for L. braziliensis showed an antileishmania activity superior to their cytotoxic effect. Although, cell permeability assays are required to make a final decision on these compounds, some knowledge on their structure-activity relationships is crucial to unravel structural modifications that maintain the activity, but reduce toxicity to human cells, or increase their lipophilicity. Considering the molecular diversity of the compounds presented in this work (modification of the substituents of the -meta and -para, addition of other lateral lipophilic residues of different sizes), compound 2b shows the best selectivity index and the highest potency against L. infantum. This results are in good agreement with the Kd value for LmPTR1, which might be related to their high sequential identity within the active site. It should also be considered that a future analysis of the PTR1 structure for the Leishmania species circulating in Brazil is something to be explored, given the diversity of species in this country and their respective epidemiological impacts.
References
Albuquerque JFC et al (1995) Synthèse et étude structurale des 5-arilidène thiazolidine-2,4-diones et 4-thio-imidazolidine-2-ones-3-substituées. Ann Pharm Fr 53:209–214
Alvar J, Arana B (2018) Leishmaniasis, impact and therapeutic needs. In: Rivas L and Gil C (ed) Drug Discov Leishmaniasis, Royal Society of Chemisty, pp 1–23
Bozdag-Dündar O et al (2007) Synthesis and antimicrobial activity of some new thiazolyl thiazolidine-2,4-dione derivatives. Bioorg Med Chem 15:6012–6017
Bruno G, Costantino L, Curinga C, Maccari R, Monforte F, Nicolò F, Ottanà R, Vigorita MG (2002) Synthesis and aldose reductase inhibitory activity of 5-Arylidene-2,4-thiazolidinedionesy. Bioorg Med Chem 10:1077–1084
Cavazzuti A, Paglietti G, Hunter WN, Gamarro F, Piras S, Loriga M, Allecca S, Corona P, McLuskey K, Tulloch L, Gibellini F, Ferrari S, Costi MP (2008) Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc Natl Acad Sci U S A 105(5):1448–1453
Cimmperman P, Matulis D (2011) Chapter 8. Protein Thermal Denaturation Measurements via a Fluorescent Dye. In: Protein thermal denaturation measurements via a fluorescent dye. Royal Society of Chemistry
Contessoto VG et al (2018) Introdução ao problema de enovelamento de proteínas: uma abordagem utilizando modelos computacionais simplificados. Rev Brasil Ensino Física 40(4):1–13
Craig DQM, Newton JM (1991) Characterisation of polyethylene glycol solid dispersions using differential scanning calorimetry and solution calorimetry. Int J Pharm 76:17–24
De Mesquita ML et al (2009) Cytotoxic activity of Brazilian Cerrado plants used in tradicional medicine against câncer cell lines. J Ethnopharmacol 123(3):439–445
Denizot F, Lang R (1986) Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 89(2):271–277
Di Giorgio C et al (2004) In vitro activity of the b-carboline alkaloids harmane, harmine, and harmaline toward parasites of the species Leishmania infantum. Exp Parasitol 106:67–74
Dupeux F, Röwer M, Seroul G, Blot D, Márquez JA (2011) A thermal stability assay can help to estimate the crystallization likelihood of biological samples. Biol Crystallogr 67:915–919
Eggert US (2013) The why and how of phenotypic small-molecule screens. Nat Chem Biol 9(4):206–209
Ericsson UB et al (2006) Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem 357:289–298
Garbett NC, Chaires JB (2012) Thermodynamic studies for drug design and screening. Expert Opin Drug Discovery 7(4):299–314
Gasteiger E et al (2005) Protein identification and analysis tools on the ExPASy Server. In: Walker JM. The Proteomics Protocols Handbook, Humana Press 571–607
Gilbert IH (2013) Drug discovery for neglected diseases: molecular target-based and phenotypic approaches. J Med Chem 56(20):7719–7726
Gontijo B, Carvalho MLR (2003) Leishmaniose tegumentar americana. Rev Soc Bras Med Trop 36(1):71–80
Gourley DG, Luba J, Hardy LW, Beverley SM, Hunter WN (1999) Crystallization of recombinant Leishmania major pteridine reductase 1 (PTR1). Acta Crystallogr Sec D 55:1608–1610
Gourley DG, Schüttelkopf AW, Leonard GA, Luba J, Hardy LW, Beverley SM, Hunter WN (2001) Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat Struct Biol 8(6):521–525
Gouveia FL, de Oliveira RMB, de Oliveira TB, da Silva IM, do Nascimento SC, de Sena KXFR, de Albuquerque JFC (2009) Synthesis, antimicrobial and cytotoxic activities of some 5-arylidene-4-thioxo-thiazolidine-2-ones. Eur J Med Chem 44:2038–2043
Grasberger BL et al (2005) Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J Med Chem 4(48):909–912
Kopec J, Schneider G (2011) Comparison of fluorescence and light scattering based methods to assess formation and stability of protein-protein complexes. J Struct Biol 175(2):216–223
Leite FHA, Santiago PBGS, Froes TQ, da Silva Filho J, da Silva SG, Ximenes RM, de Faria AR, Brondani DJ, de Albuquerque JFC, Castilho MS (2016) Structure-guided discovery of thiazolidine-2,4-dione derivatives as a novel class of Leishmania major pteridine reductase 1 inhibitors. Eur J Med Chem 123:639–648
Leprohon P, Fernandez-Prada C, Gazanion É, Monte-Neto R, Ouellette M (2015) Drug resistance analysis by next generation sequencing in Leishmania. Int J Parasitol Drugs Drug Resist 5(1):26–35
Lesyk R, Zimenkovsky B, Atamanyuk D, Jensen F, Kiec-Kononowicz K, Gzella A (2006) Anticancer thiopyrano[2,3-d][1,3]thiazol-2-ones with norbornane moiety. Synthesis, cytotoxicity, physico-chemical properties, and computational studies. Bioorg Med Chem 14:5230–5240
Libermann D, Himbert J, Hengl L (1948) La thiazolidione, point de depart d’une synthèse des acides thiopyruviques et thioglyoxiliques substituèes. Bull Soc Chim Fr 11–12:1120–1124
Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, Ellestad G (2004) Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem 332:153–159
Nare B, Hard LW, Beverley SM (1997) The roles of pteridine reductase 1 and synthase in pteridine metabolism in the protozoan parasite Leishmania major. J Biol Chem 272(21):13883–13891
Nare B, Garraway LA, Vickers TJ, Beverley SM (2009) PTR1-dependent synthesis of tetrahydrobiopterin contributes to oxidant susceptibility in the trypanosomatid protozoan parasite Leishmania major. Curr Genet 55:287–299
Niesen FH, Berglund H, VEdadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2(9):2212–2221
Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR (2001) High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen 6(6):429–440
Patra N, de U, Kang JA, Kim JM, Ahn MY, Lee J, Jung JH, Chung HY, Moon HR, Kim HS (2011) A novel epoxypropoxy flavonoid derivative and topoisomerase II inhibitor, MHY336, induces apoptosis in prostate cancer cells. Eur J Pharmacol 658:98–107
Pereira JR et al (2012) Cytotoxicity evaluation of marine alkaloid analogues of viscosaline and theonelladin C. Biomed Prev Nutrit 2:145–148
Petrauskas V et al (2019) Inhibitor binding to carbonic anhydrases by fluorescent thermal shift assay. In: Matulis D (ed) Carbonic anhydrase as drug target: thermodynamics and structure of inhibitor binding, Springer, pp 63–78
Pikkemaat MG, Linssen ABM, Berendsen HJC, Janssen DB (2002) Molecular dynamics simulations as a tool for improving protein stability. Protein Eng 15:185–192
Priest B, Erdemli G (2014) Phenotypic screening in the 21st century. Front Pharmacol 5(264):1–2
Sancho J (2013) The stability of 2-state, 3-state and more-state proteins from simple spectroscopic techniques... plus the structure of the equilibrium intermediates at the same time. Arch Biochem Biophys 531:4–13
Schoonen WGEJ, Westerink WM, de Roos JA, Débiton E (2005) Cytotoxic effects of 100 reference compounds on Hep G2 and HeLa cells and of 60 compounds on ECC-1 and CHO cells. I Mechanistic assays on ROS, glutathione depletion and calcein uptake. Toxicol in Vitro 19:505–516
Sienkiewicz N, Ong HB, Fairlamb AH (2010) Trypanosoma brucei pteridine reductase 1 is essential for survival in vitro and for virulence in mice. Mol Microbiol 77(3):658–671
Sievers F et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7(539):1–6
Silva IM et al (2014) Synthesis and antimicrobial activities of 5-arylidene-thiazolidine-2,4-dione derivatives. Biomed Res Int 2014:1–8
Stockert JC, Blázquez-Castro A, Cañete M, Horobin RW, Villanueva Á (2012) MTT assay for cell viability: intracellular localization of the formazan product is in lipid droplets. Acta Histochem 114(8):785–796
Swinney DC, Anthony J (2011) How were new medicines discovered? Nat Rev Drug Discov 10(7):507–519
Teixeira BVF et al (2019) Dual and selective inhibitors of pteridine reductase 1 (PTR1) and dihydrofolate reductase-thymidylate synthase (DHFR-TS) from Leishmania chagasi. J Enzyme Inhib Med Chem 27(6):1439–1450
Teles ALB et al (2018) Identification, characterization and molecular modelling studies of Schistosoma mansoni dihydrofolate reductase inhibitors: from assay development to hit identification. Curr Top Med Chem 18:1–10
Vicini P, Geronikaki A, Anastasia K (2006) Synthesis and antimicrobial activity of novel 2-thiazolylimino-5-arylidene-4-thiazolidinones. Bioorg Med Chem 14:3859–3864
Vickers TJ, Beverley SM (2011) Folate metabolic pathways in Leishmania. Essays Biochem 51:63–80
Vidal LS et al (2018) Review of NAD(P)H-dependent oxidoreductases: properties, engineering and application. BBA Proteins Proteom 1866(2):327–347
Vivoli M et al (2014) Determination of protein-ligand interactions using differential scanning fluorimetry. J Vis Exp 91:1–13
Wagner BK, Schreiber SL (2016) The power of sophisticated phenotypic screening and modern mechanism-of-action methods. Cell Chem Biol 23(1):3–9
Waldron TT, Murphy KP (2003) Stabilization of proteins by ligand binding: application to drug screening and determination of unfolding energetics. Biochemistry 42:5058–5064
World Health Organization (2017) http://www.who.int/gho/neglected_diseases/leishmaniasis/en/. Accessed 04 Set 2017
World Health Organization. Leishmaniasis (2019) https://www.who.int/news-room/fact-sheets/detail/leishmaniasis. Accessed 12 Abr 2019
Zaia DAM, Zaia CTBV, Lichtig J (1998) Determinação de proteínas totais via espectrofotometria: vantagens e desvantagens dos métodos existentes. Química Nova 21(6):787–793
Zhang R, Monsma F (2010) Fluorescence-based thermal shift assays. Curr Opin Drug Discov Dev 13(4):389–402
Funding
I would like to thank FAPESB and CNPq (479160/2013-9) for the financial support to carry out this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Section Editor: Ramaswamy Kalyanasundaram
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Neri, F.S.M., Júnior, D.B.C., Froes, T.Q. et al. Antileishmanial activity evaluation of thiazolidine-2,4-dione against Leishmania infantum and Leishmania braziliensis. Parasitol Res 119, 2263–2274 (2020). https://doi.org/10.1007/s00436-020-06706-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-020-06706-3