
Abstract
Recent studies have revealed extensive genetic variations among Neospora caninum, a cyst-forming protozoan parasite that is one of the main causes of bovine abortion in the cattle industry worldwide. Previous genetic studies based on multilocus microsatellite genotyping (MLGs) of different Ibero-American populations showed a high genetic diversity. These studies provided clear clues of a predominant clonal propagation in cattle and population sub-structuring partially associated with geographical origin. Although, these reports were limited to a reduced number of countries. In this study, the N. caninum isolates from aborted bovine fetuses and stillbirths and a goat abortion from Northern Italy were investigated genetically using 9 microsatellite markers. Complete or nearly complete isolate profiles were obtained from 30 fetuses and stillbirths. An extensive genetic diversity was also found in this Italian N. caninum population. The study of genetic relationships among Italian MLGs using network (eBURST) and principal component analyses based on the allele-sharing coefficient (PCoA) showed different clonal subpopulations disseminated throughout Northern Italy without apparent segregation depending on the geographic origin, cattle breed, or time of collection. The presence of linkage disequilibrium supports a predominant clonal propagation of Italian N. caninum. In addition, most of Italian MLGs segregated from other global populations including Spain, Argentina, Mexico, Brazil, Germany, and Scotland, suggesting the existence of specific N. caninum subpopulations in the Northern Italy and different subpopulations of N. caninum circulating in Europe.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neospora caninum is an obligate intracellular parasite that causes abortions, neonatal death, and stillbirths in cattle (Dubey and Schares 2011). Cattle act as intermediate hosts and can become infected via the ingestion of oocysts (horizontal transmission) shed by the definitive host (canids) after sexual reproduction. In addition, N. caninum is transmitted transplacentally in cattle very efficiently. Neospora caninum is distributed worldwide and has been associated with considerable economic losses in the cattle industry (Reichel et al. 2013).
There is evidence that N. caninum isolates differ in virulence that may explain variations in disease epidemiology and clinical presentation in the field (Dubey and Schares 2011). There is also knowledge on genetic diversity among isolates that suggests the presence of genetically different populations in N. caninum. In the absence of described single nucleotide and insertion-deletion polymorphisms suitable for genotyping, microsatellite markers (MS) have been broadly used to study genetic diversity in N. caninum (Regidor-Cerrillo et al. 2006; Al-Qassab et al. 2009; Regidor-Cerrillo et al. 2013; Medina-Esparza et al. 2016). These studies revealed the presence of great genetic diversity worldwide with different N. caninum populations in countries from the Americas (Argentina and Mexico) and Europe (Spain, Germany, and Scotland) (Regidor-Cerrillo et al. 2013, Medina-Esparza et al. 2016). Genotyping with these markers demonstrated population sub-structuring partially associated with the geographical origin. Interestingly, the closest genetic relationships were established between Ibero-American (Spanish and Argentinean or Mexican) populations, even more than among European populations.
In this work, we studied the population of N. caninum circulating in bovine abortions and stillbirths and a goat abortion from the Northern Italy. More than the 70% of the national assets of cattle production are located in the Northern Italy, primarily in Lombardy (27%), Piedmont (13%), Veneto (15%), and Emilia-Romagna (10%), under intensive farming system, but extensive pastures are also typical of the Alps region. Dairy cattle farming in Italy is mainly based on the Holstein breed originating from Germany and Holland, but genetically very similar throughout the European countries. Some other local breeds, such as Rendena or Bruna, are mainly farmed in the Alps area. Cattle beef farming in Italy includes many breeds, both imported and autochthonous. The imported breeds are mainly from France (Charolaise and Limousine), imported as calves, fattened and slaughtered in a few months. The most important, represented, and the renewed autochthonous breed distributed in Northern Italy is the Piedmontese. The overall seroprevalence of N. caninum in Northern Italy beef and dairy herds was determined to be 50.5% and the individual seroprevalence was 16% (Otranto et al. 2003). The seroprevalence of N. caninum infection was 19% in the Piedmontese breed (Almería et al. 2009).
We investigated genetic relationships of the North Italian N. caninum isolate genotypes with other isolate populations described in previous studies of Spain, Argentina, Mexico, Brazil, Germany, and Scotland (Regidor-Cerrillo et al. 2013; Medina-Esparza et al. 2016).
Material and methods
A total of 40 brain samples from aborted bovine fetuses and stillbirths from the Piedmont (Northwestern Italy, PMN), Lombardy (North-Central Italy, LOM), and Veneto-Trento province (Northeastern Italy, VEN) were included in this study (Table 1). In addition, one sample obtained from a goat fetus abortion was included in this study. Samples were collected between 2013 and 2016 from clinical cases submitted to the Istituto Zooprofilattico Sperimentale delle Venezie for differential diagnosis. The samples from Piedmont and Lombardy were both from beef Piedmontese cattle and dairy cattle (Holstein), while the samples from the Veneto region were from Holstein, but also originated from crossbreed cattle or local dairy cattle breeds such as Rendena (Table 1).
Brain samples were collected from all abortion and stillbirth cases and maintained at − 20 °C until used for DNA extraction. DNA was extracted from approximately 40 mg of each sample using “High Pure PCR Template Preparation Kit” (Roche Diagnostics), according to the manufacturer’s instructions. A coccidian specific RFLP-PCR assay, targeting the small subunit of 18S rRNA gene because of its low intraspecific variability, was applied to all of the DNA samples as described by Ho et al. (1996) with minor modifications. Briefly, the PCR reaction mixture (40 μL) contained 4 μL of DNA (50–100 ng), 1× DNA polymerase reaction buffer, 2 μM of dNTPs, 1 mM of magnesium chloride, 1 M of betaine, 1 U of FastStart Taq DNA polymerase (Roche Diagnostics), and 1 mM of each primer APIF-1 (5′-AAG TAT AAG CTT TTA TAC GGC-3′) and APIR-1 (5′-CAC TGC CAC GGT AGT CCA ATA-3′) final concentration. Amplification thermal conditions were as follows: hot start step at 95 °C for 15 min, followed by 10 cycles of denaturation step at 94 °C for 30 s, annealing at 54 °C for 1 min, extension at 72 °C for 2 min and 30 more cycles of denaturation step at 94 °C for 30 s, annealing at 52 °C for 1 min, extension at 72 °C for 2 min, followed by a final prolonged extension step of 10 min at 72 °C. All amplicons were visualized using 7% acrylamide gel electrophoresis stained with silver nitrate. Positive amplicons of 305 base pairs were then subjected to enzymatic restriction analysis (RE) with BseDI Fast (Fermentas) at 60 °C for 90 min. A specific N. caninum restriction pattern of 3 fragments (85, 92, and 105 bp) was observed in positive samples using gel electrophoresis and staining as described above.
Neospora caninum–positive DNA samples were used for genotyping by multilocus microsatellite analysis using 9 MS previously described (Regidor-Cerrillo et al. 2013). Specifically, MS4, MS5, MS6A, MS6B, MS7, MS8, MS10, MS12, and MS21 markers were amplified using specific primers under nested PCR conditions and analyzed as described by Regidor-Cerrillo et al. (2013). The sizes of the PCR products for all of the microsatellites were determined in a 48-capillary 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA) with GeneScan-500 (LIZ) size standards (Applied Biosystems), and MS7 and MS10 markers were sequenced using Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) and a 3730 DNA Analyzer (Applied Biosystems) at the Genomics Unit of the Complutense University of Madrid. Allele assignment and microsatellite multilocus genotypes (MLGs) were numbered according to a previous dataset (Regidor-Cerrillo et al. 2013; Medina-Esparza et al. 2016).
Genetic relationships among the N. caninum microsatellite multilocus genotypes were estimated by eBURST and principal coordinates analysis (PCoA). The eBURST software (Feil et al. 2004) and GenAlEx 6.502 package (Peakall and Smouse 2012), respectively, were used to explore the genetic relationships within and among Italian MLGs and the entire dataset with 100 MLGs including Spanish (49), Argentinean (13), Mexican (10), German (8), Scottish (7), and Brazilian (3) MLGs (see supplementary Table S1). MLGs from 11 N. caninum isolates that had been obtained from different geographical locations, including Italy and Brazil, were considered a worldwide group of references and out-groups for comparisons (Regidor-Cerrillo et al. 2013). The eBURST software generates networks composed of MLGs represented as dots, whose diameter is proportional to the number of identical genotypes, linked to their single-locus (SLV-8 shared loci of 9) and double-loci (DVL-7 shared loci of 9) variants by lines. As the missing data are not identified, MLGs with nearly complete allelic data were included by assigning a mock allele for the missing MS. Also, a pairwise, individual-by-individual, genetic distance matrix using the allele-sharing coefficient was used to generate a covariance matrix and perform a PCoA. Nearly completed MLGs were included in the analysis under the “interpolate missing alleles” option. Nei’s genetic unbiased distance (Nei 1978) was also estimated among geographical N. caninum populations using GenAlEx 6.503 package. Multilocus linkage disequilibrium (LD) among complete Italian MLGs was also assessed using a standardized index of association (IAS), implemented in LIAN v3.7 web interface (Haubold and Hudson 2000; http://guanine.evolbio.mpg.de/cgi-bin/lian/lian.cgi.pl/query). The presence of LD was assessed for whole Italian and Piedmont and Veneto populations, while the Lombardy population was barely represented from a statistical point of view.
Results
Identification of Italian microsatellite multilocus genotypes and population analysis
Microsatellite amplification was achieved from 40 of 41 samples and only failed from a unique DNA sample. Complete (n = 24) or nearly complete MLGs (n = 6), with at least 8 microsatellites identified, were obtained from 30 out of 40 samples (Table 1). New alleles were identified for the MS7 and MS10 markers. Within the MS7 marker, two types of alleles were previously described: one group based on TA repeat and a second group carrying a single nucleotide polymorphism (SNP) − 2 bp from the TA repetitive motif that results in an additional TA and at present with only two alleles previously identified, the 9.1 and the 10.1 (Table S1). A new allele was identified in this study for each group: one based on 17 TA repeats and the allele 8.1 (Table 1). A high degree of polymorphism was found in the MS7 locus from the Italian population with 10 different alleles out of 31 samples amplified (Table 1). In addition, 14 new alleles were identified in the MS10 marker (Table 1). MS10 was the most polymorphic with 23 identified different alleles out of the amplified 34 DNA samples.
When examining complete or nearly complete MLGs, two samples (ITA-14-14 and ITA-15-30) with different geographical origins and years of collection showed identical MLGs. Also, nearly complete MLG of the sample ITA-14-15 shared all identified markers with these MLGs and originated from the same herd as the ITA-14-14 sample. In addition, ITA-15-33 and the nearly complete ITA-15-35 that were collected from the same herd and year shared identified MS alleles. The remaining samples showed unique MLGs. Mean genetic diversity (He) was assessed high (0.7061 ± 0.0969) for this Italian population.
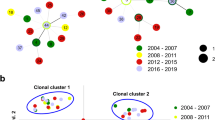
The eBURST analysis with complete and nearly complete MLGs was carried out to reveal clusters with the closest genetic relationship among Italian isolates. Three clonal DLV groups (with n ≥ 3 MLGs) were found connecting 6, 5, and 3 MLGs (Fig. 1a). A clonal group of 6 MLGs was clustered closest MLGs from the same herd ITA-15-35 and ITA-15-33, as previously described, with other MLGs without clear association by geographical location, or from beef or dairy breeds, or year of collection of sample (Fig. 1a, Table 1, group 1). ITA-14-14/ITA-15-30 MLG and the ITA-14-15 MLG also appeared connected with the ITA-14-18 MLG, grouping N. caninum samples from the Piedmont, Lombardy, and Veneto regions (Fig. 1a; Table 1, group 3). Additionally, PCoA using the complete and nearly complete of the Italian dataset showed non-segregation of MLGs according to geographical origin or year of collection. The second principal component accounted for 35% of variation with a distribution of MLGs from the Piedmont and Veneto regions by three different quadrants. Genetic clusters recognized by eBURST were identified and located in three different quadrants (Fig. 1b). The goat genotype (ITA-16-41) was not associated with any cluster.

Genetic relationships among the Neospora caninum Italian MLGs estimated by eBURST (a) and PCoA (b) analyses. a In the eBURST analysis, each MLG is represented by a colored dot according to their geographical origin (PMN, Piedmont; LOM, Lombardy; and VEN, Veneto-Trento; see legend). The dot shape represents the year of sample collection (see legend). Single-locus variants (SLV) are connected by purple lines and double-loci variants (DLV) by blue lines. The main MLG clusters (n ≥ 3) are represented. Singleton and MLG clusters ≤ 3 were excluded from the snapshot representation. b Graphics represent the genetic relationships between the Italian MLGs based on a covariance matrix. A pairwise, individual-by-individual, genetic distance matrix using the allele-sharing coefficient was used to generate a covariance matrix and perform a principal coordinates analysis (PCoA) using the GenAlEx 6.502 package (Peakall and Smouse 2012). The colors indicate the geographic origin (PMN, Piedmont; LOM, Lombardy; and VEN, Veneto-Trento; see legend). The proportion of the variation in the dataset for coordinate 1 was 20.32% (x axis), and that for coordinate 2 (y axis) was 14.74%
Allele variations detected among the MLGs inside all of the detected clusters are reported in Table 1. The group of 3 MLGs conserved most of the identified alleles without variations being limited to MS10 change in the ITA-14-18 MLG (group 3). Polymorphism was detected in a limited number of MSs (2 to 3) inside the other 2 groups with alleles differing only in 1 or 2 repeat units indicating linkage between loci. A group of 6 MLGs showed variation in MS4, MS6A, and MS10 markers (group 1) and a group of 5 MLGs in their MS5 and MS7 markers (group 2). The level of LD was also estimated using the IAS for whole Italian dataset at the first level and the Piedmont and Veneto-Trento populations at the second level. The value of IAS was positive for the whole Italian dataset and Piedmont region, indicating LD (see supplementary Table S2). The Veneto-Trento population showed a positive value of IAS but close to 0, denoting absence of LD, likely due to the limited number of MLGs (n < 10) (Table S2).
Genetic relationships with N. caninum isolates from other countries
The relationship between complete Italian MLGs with other complete MLGs from different geographical locations was also examined using eBURST (supplementary Fig. S1). Five clusters (n ≥ 3 MLGs) were recognized. The major cluster consisted of 43 MLGs, including those from Spain (n = 27), Argentina (n = 5), Brazil (two Brazilian and the Nc-Bahia MLG of the worldwide group; n = 3), Mexico (n = 2), Germany (n = 1), and Italy (n = 5) (group G1; Fig. S1). The Italian MLGs (n = 5) that clustered in this main group included MLGs of group 2 previously identified in the eBURST analysis of the Italian population (Fig. 1a, Table 1, group 2). The second group associated five MLGs distributed throughout Europe without apparent geographical connection and included two Scottish MLGs, two German MLGs (one German and the Hh-Berlin MLG from the worldwide group), and one Spanish MLG (group G2; Fig. S1). The third group connected four Argentinean MLGs (group G3; Fig. S1). Finally, the last two groups linked two and three Italian MLGs with one Spanish and one from the UK (the Nc-Liv isolate from the worldwide group), respectively (group G4 and group G5; Fig. S1). These last two groups clustered Italian MLGs with direct association in the previous eBURST analysis when the Italian population was independently analyzed (Fig. 1a, Table 1, groups 1 and 3).
The genetic relationships of the Italian MLGs together with Spanish, Argentinean, Mexican, Brazilian, German, and Scottish N. caninum MLGs were also re-examined by an overall PCoA using the complete and nearly complete MLG dataset (Fig. 2a). The two axes accounted for 21.2% of the variations, showing that the geographical populations were not entirely segregated. Nevertheless, the majority of Italian MLGs (n = 21 out of 30) were distributed among the far left quadrants, together with the majority of the German (n = 6 out of 8) and Scottish (n = 5 out of 7) MLGs. Moreover, the majority of the Spanish MLGs (n = 38 out of 56) were distributed among the far right quadrants, together with approximately 50% of the Argentinean (n = 7 out of 13) and Mexican (n = 5 out of 11), and all of the Brazilian (n = 3) MLGs. A new PCoA according to the mean genetic distance among the geographical populations showed clear differentiation of the Ibero-American populations in the far right quadrants from the European populations. The German and Italian populations were segregated from the Scottish population by the second axis (Fig. 2b).

Genetic relationships among the Neospora caninum MLGs from different counties estimated by PCoA. a Graphic represent the genetic relationships among MLGs based on a pairwise, individual-by-individual, genetic distance matrix using the allele-sharing coefficient to generate a covariance matrix and perform a principal coordinates analysis (PCoA) using the GenAlEx 6.502 package (Peakall and Smouse 2012). The colors indicate the geographic origin (AM, America; GER, Germany; ITA, Italy; SCO, Scotland; and SPA, Spain; see legend). The Argentinean, Brazilian, and Mexican MLGs were grouped under American origin, and the worldwide MLGs from the reference group were not included to simplify the representation. The proportion of the variation in the dataset for coordinate 1 was 13.85% (x axis), and that for coordinate 2 (y axis) was 7.37%. b Graphic represent the genetic relationships among the geographical N. caninum populations according to the mean genetic distance. The colors indicate geographic origin (ARG, Argentina; BRA, Brazil; GER, Germany; ITA, Italy; MEX, Mexico; SCO, Scotland; and SPA, Spain). The proportion of the variation in the dataset for coordinate 1 was 53.76% (x axis), and that for coordinate 2 (y axis) was 27.04%
Population differentiation was also estimated between the geographic regions using Nei’s unbiased genetic distance (Nei 1978). Nei’s unbiased genetic distance analyses confirmed a higher genetic differentiation between the Italian and the Ibero-American populations (Spanish, Argentinean, Brazilian, and Mexican populations). However, the Italian population was genetically closer to the German and Scottish populations (Table 2).
Discussion
In this study, a panel of N. caninum isolates from bovine abortions and stillbirths and a goat abortion in the Northern Italy over a 4-year period of time was genotyped using nine polymorphic markers to define the MLGs. The MLG analysis showed a high level of diversity within the Italian N. caninum isolates, with 23 different MLGs identified in the 24 samples successfully genotyped for the 9 MS. The extensive genetic diversity found in the Northern Italy was similar to what has been previously reported in other N. caninum country populations (Regidor-Cerrillo et al. 2013).
The study of relationships among the Italian MLGs showed some degree of sub-structuration in the parasite population. The eBURST and PCoA analyses showed clustering/segregation of MLGs in at least three clusters (or subpopulations) grouping approximately 50% of the Italian MLGs. Clustering was in the absence of an association to the time of collection or geographical area. Moreover, identical MLGs were found in samples from two herds geographically very distant and collected in different years. To date, the detection of identical MLGs was restricted to samples from the same herd (Regidor-Cerrillo et al. 2013; Medina-Esparza et al. 2016). Allele variations between connected MLGs in the detected clusters were in a limited number of MS markers located on different chromosomes and based only in a single/double repeat unit changes that can happen under clonal propagation in the field (González-Warleta et al. 2018). These findings suggest high spreading by clonal propagation of at least three different predominant subpopulations throughout the Northern Italy. Notwithstanding additional Italian MLGs remained without clustering, possibly representing sexual recombination or members of new divergent clonal clusters. The Italian N. caninum isolates and the Piedmont subpopulation, including exclusively single MLGs, remained in LD, indicating once more that there was a limited genetic exchange. Thus, the observed results in the Italy N. caninum population are consistent with the predominance of clonal propagation under genetic divergence by mutation of these highly polymorphic markers as was suggested in other previously studied N. caninum populations (Regidor-Cerrillo et al. 2013). This clonal model would be compatible with the life cycle of N. caninum, in which the parasite is predominantly transmitted by vertical transmission during pregnancy to successive generations and horizontal transmission could happen but with reduced probabilities of breeding of different isolates in the definitive host. New investigations based on complete genome sequencing of different isolates support the clonal model as the main propagation of N. caninum globally from its separation from its ancestors (Khan et al. 2019).
Under a predominance of clonal propagation of N. caninum, the dissemination of clonal clusters could be a consequence of animal movement within the region. The majority of MLGs were identified on samples from Holstein breed cattle. Italian Holsteins are derived from Dutch and North American Holstein breeds imported to Italy in the late twentieth century and is currently the most common dairy breed (Mancini et al. 2014). Two out of the three Italian N. caninum clonal clusters exclusively grouped genotypes from Holstein cattle (Table 1, group 2 and group 3). Interestingly, the greatest cluster (group 1, Table 1) grouped MLGs from Holstein and Piedmontese breeds. Until recently, Piedmontese cattle breeding has been restricted to the region of origin of Piedmont (Mancini et al. 2014). During the last 20 years, Piedmontese cattle have been extended throughout Northern Italy and have been introduced into other distant locations, such as Canada, USA, and New Zealand. These findings indicate that there is an absence of clear MLG segregation by cattle breed, suggesting the circulation of these N. caninum isolates by the route of horizontal transmission between breeds. A minor role has been attributed to the horizontal transmission of N. caninum in cattle, although this route could be relevant in some scenarios (Dubey and Schares 2011). However, we cannot determine the origin of these genotypes shared by different breeds. The Piedmontese cattle are characterized by a muscular hypertrophy, commonly known as “double muscling,” a trait desirable in beef cattle. Over the last 50 years, the Piedmontese breed has been strongly selected for the double muscling selecting homozygous cattle for this trait (Mancini et al. 2014). This could be translated to a “bottleneck” diminishing diversity of N. caninum or N. caninum infection in Piedmontese cattle. We may assume the introduction of N. caninum genotypes via Holstein breed as the most likely event, although additional studies are necessary.
The genetic relationship of the Italian N. caninum isolates with those in other countries was also investigated. Two of the three identified clonal subpopulations were segregated independently from other N. caninum isolates with different geographical origin (Table 1, group 1 and group 3). These two subpopulations may have originated from Italy that genetically differed, although new studies from other geographical regions and breeds are necessary to confirm this hypothesis. The third subpopulation was genetically associated with a common cluster with Ibero-American MLGs (Table 1, group 2). This cluster was the principal group formed by a large number of MLGs from the Spanish population identified in previous studies (Regidor-Cerrillo et al. 2013). It was previously hypothesized that genetic relationships among Ibero-American N. caninum populations may reflect a common origin via the introduction of Iberian cattle in Latin America (Regidor-Cerrillo et al. 2013). In this study, the number of Italian MLGs associated with this main cluster was approximately 20% of the isolates. Despite this reduced number of MLGs, it is tempting to infer that this cluster may be a N. caninum population distributed throughout the Americas and Europe, although it is a minority in Northern Italy. The Holstein breed has been broadly extended during the past few decades and can now be found on every continent and in almost every country, which may support the expansion of N. caninum for this cluster. Nevertheless, N. caninum MLGs from Holstein cattle from Germany failed to be clustered in this main N. caninum cluster; although interestingly, the Italian and German populations showed the closest genetic relationship. However, the Italian N. caninum population diverged genetically from the Spanish and other Ibero-American populations, typical of geographically disconnected populations in the absence of migration. Germany and Italy are the nearest geographically, and the interchange of animals between these countries may be reflected in these findings. Nevertheless, the limited number of German MLGs hampers the interpretation of the results.
In summary, this study’s findings demonstrate a high genetic diversity of N. caninum involved in the reproductive failure of bovines and the existence of different N. caninum subpopulations possibly throughout the Northern Italy under clonal spreading by vertical and horizontal transmission. The Italian N. caninum subpopulations differed genetically from other American and European populations, suggesting specificity of N. caninum from Northern Italy and different subpopulations circulating in Europe. As there are differences in virulence within N. caninum isolates, the population diversity circulating in Europe may be implicated on the observed differences in the prevalence, transmission, and occurrence of disease in different countries (Dubey and Schares 2011). New studies will be necessary to discern whether there is a link between genotype and virulence, at least, at the local level and evaluate how knowledge of genotype may be used to control neosporosis on the farm.
References
Almería S, López-Gatius F, García-Ispierto I, Nogareda C, Bech-Sàbat G, Serrano B, Santolaria P, Yániz JL (2009) Effects of crossbreed pregnancies on the abortion risk of Neospora caninum-infected dairy cows. Vet Parasitol 163:323–329. https://doi.org/10.1016/j.vetpar.2009.04.026
Al-Qassab S, Reichel MP, Ivens A, Ellis JT (2009) Genetic diversity amongst isolates of Neospora caninum and the development of a multiplex assay for the detection of distinct strains. Mol Cell Probes 23:132–139
Dubey JP, Schares G (2011) Neosporosis in animals-the last five years. Vet Parasitol 180:90–108. https://doi.org/10.1016/j.vetpar.2011.05.031
Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG (2004) eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol 186:1518–1530. https://doi.org/10.1128/jb.186.5.1518-1530.2004
González-Warleta M, Castro-Hermida JA, Calvo C, Pérez V, Gutiérrez-Expósito D, Regidor-Cerrillo J, Ortega-Mora LM, Mezo M (2018) Endogenous transplacental transmission of Neospora caninum during successive pregnancies across three generations of naturally infected sheep. Vet Res 49:106. https://doi.org/10.1186/s13567-018-0601-3
Haubold B, Hudson RR (2000) LIAN 3.0: detecting linkage disequilibrium in multilocus data. Linkage Analysis. Bioinformatics. 16:847–848. https://doi.org/10.1093/bioinformatics/16.9.847
Ho MSY, Barr BC, Marsh AE, Anderson ML, Rowe JD, Tarantal AF, Hendrickx AG, Sverlow K, Dubey JP, Conrad PA (1996) Identification of bovine Neospora parasites by PCR amplification and specific small-subunit rRNA sequence probe hybridization. J Clin Microbiol 34:1203–1208
Khan A, Fujita AW, Randle N, Regidor-Cerrillo J, Shaik JS, Shen K, Oler AJ, Quinones M, Latham SM, Akanmori BD, Cleaveland S, Innes EA, Ryan U, Šlapeta J, Schares G, Ortega-Mora LM, Dubey JP, Wastling JM, Grigg ME (2019) Global selective sweep of a highly inbred genome of the cattle parasite Neospora caninum. Proc Natl Acad Sci U S A In press doi. https://doi.org/10.1073/pnas.1913531116
Mancini G, Gargani M, Chillemi G, Nicolazzi EL, Marsan PA, Valentini A, Pariset L (2014) Signatures of selection in five Italian cattle breeds detected by a 54K SNP panel. Mol Biol Rep 41:957–965. https://doi.org/10.1007/s11033-013-2940-5
Medina-Esparza L, Regidor-Cerrillo J, García-Ramos D, Álvarez-García G, Benavides J, Ortega-Mora LM, Cruz-Vázquez C (2016) Genetic characterization of Neospora caninum from aborted bovine foetuses in Aguascalientes. Mexico Vet Parasitol 228:183–187. https://doi.org/10.1016/j.vetpar.2016.09.009
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics. 89:583–590
Otranto D, Llazari A, Testini G, Traversa D, Frangipane di Regalbono A, Badan M, Capelli G (2003) Seroprevalence and associated risk factors of neosporosis in beef and dairy cattle in Italy. Vet Parasitol 118:7–18. https://doi.org/10.1016/j.vetpar.2003.10.008
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research--an update. Bioinformatics. 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Regidor-Cerrillo J, Pedraza-Díaz S, Gómez-Bautista M, Ortega-Mora LM (2006) Multilocus microsatellite analysis reveals extensive genetic diversity in Neospora caninum. J Parasitol 92:517–524. https://doi.org/10.1645/GE-713R.1
Regidor-Cerrillo J, Díez-Fuertes F, García-Culebras A, Moore DP, González-Warleta M, Cuevas C, Schares G, Katzer F, Pedraza-Díaz S, Mezo M, Ortega-Mora LM (2013) Genetic diversity and geographic population structure of bovine Neospora caninum determined by microsatellite genotyping analysis. PLoS One 8:e72678. https://doi.org/10.1371/journal.pone.0072678
Reichel MP, Ayanegui-Alcérreca A, Gondim LFP, Ellis J (2013) What is the global economic impact of Neospora caninum in cattle–the billion dollar question. Int J Parasitol 43:133–142. https://doi.org/10.1016/j.ijpara.2012.10.022
Funding
This project was financed by the Community of Madrid and co-financed by the European Regional Development Fund and the European Social Fund (PLATESA2-CM P2018/BAA-4370).
Author information
Authors and Affiliations
Contributions
NA, ES, and LC conducted the sampling and PCR detection, and data collection. JRC and PH carried out sample genotyping and genetic analyses. JRC and PH interpreted the genotyping data. JRC, PH, and LMOM wrote the paper. NA, LC, and ES discussed and commented the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Section Editor: Larissa Howe
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Table S1
Multilocus microsatellite genotyping database including the Argentinean, Brazilian, Italian, Mexican, Spanish, Scottish and German MLGs was used for the genetic relationship analyses in this study. (DOCX 37 kb)
Table S2
Linkage equilibrium and disequilibrium in Italian Neospora caninum populations (DOCX 18.3 kb)
Fig. S1
Relationships among the Neospora caninum MLGs estimated by the eBURST analysis. Each complete MLG is represented by a dot. MLG dots were also colored according to their geographical origin (see legend). The dot diameter is proportional to the number of samples with identical MLG (see legend). Single locus variants (SLV) are connected by purple lines and double loci variants (DLV) by blue lines. MLGs clusters (n>3 MLGs) are represented: Group 1 to 5. Singletons were excluded from the snapshot representation. (PPTX 346 kb)
Rights and permissions
About this article

Cite this article
Regidor-Cerrillo, J., Horcajo, P., Ceglie, L. et al. Genetic characterization of Neospora caninum from Northern Italian cattle reveals high diversity in European N. caninum populations. Parasitol Res 119, 1353–1362 (2020). https://doi.org/10.1007/s00436-020-06642-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-020-06642-2