
Abstract
Intra-leukocytic gamonts consistent with the description of Hepatozoon griseisciuri Clark, 1958 are reported for the first time in Canadian eastern gray squirrels (Sciurus carolinensis Gmelin, 1788). Polymerase chain reaction (PCR) amplification and direct Sanger sequencing identified a pair of distinct genotypes at both a nuclear and mitochondrial locus; two 18S ribosomal RNA gene sequences (rDNA; genotype A and genotype B: 1816 base pairs (bp); 98.8% pairwise identity) and 2 distinct complete mitochondrial genome sequences (genotype A: 6311 bp; genotype B: 6114 bp; 89.1% pairwise identity) were obtained from 3 H. griseisciuri-infected squirrels sampled in Guelph, Ontario. The genetic content of both circular-mapping mitochondrial genomes was conventional for apicomplexan protists; each encoded for 3 protein-coding genes (cytochrome c oxidase subunit I (COI); cytochrome c oxidase subunit III (COIII); and cytochrome B (CytB)), 14 fragmented large subunit rDNA, 10 fragmented small subunit rDNA, and 8 unassigned rDNA. These genotypes, based on sequences obtained from a pair of loci from two parasite genomes, confirm the presence of at least two Hepatozoon species infecting Ontario eastern gray squirrels, one of which is likely to be conspecific with H. griseisciuri.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus Hepatozoon (Adeleorina) was erected by Miller (1908) to support the description of Hepatozoon perniciosum Miller, 1908 (later synonymized as Hepatozoon muris (Balfour, 1906) Wenyon, 1926), an apicomplexan parasite of the rodent mite Laelaps (subgenus Echinolaelaps) echidnina Berlese, 1887 that utilized the leukocytes of laboratory rat intermediate hosts. The genus Hepatozoon later expanded to include species infecting a vast range of invertebrate definitive hosts (e.g., various dipteran insects, ticks, mites, fleas, lice) and vertebrate intermediate hosts (e.g., mammals, amphibians, reptiles, birds) (Smith 1996; Barta 2000). Given the breadth of hosts associated with Hepatozoon spp., the monophyly of the genus has been put into question (Smith and Desser 1997; Karadjian et al. 2015).
Hepatozoon species have been reported from a number of different sciurid hosts (see Clark 1958) but Hepatozoon griseisciuri Clark, 1958 is currently the only Hepatozoon species described in the eastern gray squirrel, Sciurus carolinensis Gmelin, 1788. First observed in the leukocytes of the eastern gray squirrel, Hermann and Price (1955) identified the gamonts as Hepatozoon sciuri based on Coles’ (1914) description of a morphologically similar parasite from a Eurasian red squirrel (Sciurus vulgaris) in the UK. Based on the description of additional lifecycle stages, Clark (1958) named H. griseisciuri and determined the definitive host of H. griseisciuri to be a mite commonly identified at that time as Euhaemogamasus ambulans (Thorell, 1872); Redington (1970) later concluded in a comprehensive taxonomic revision that the correct identification of this mite definitive host was Haemogamasus reidi Ewing, 1925. Clark (1958) concluded that H. griseisciuri could “be distinguished from all other adequately known species of Hepatozoon by the fact that the sporocysts regularly contain only 4 large sporozoites.” In addition, Clark (1958) found that sporogonic development was possible in laboratory-reared spiny rat mites, L. echidnina, that are not typically found on eastern gray squirrels. Hepatozoon griseisciuri has a two-host lifecycle starting in the squirrel as merogonic replication in the tissues (e.g., lung, spleen, liver, bone marrow) followed by gamonts that mature in leukocytes (Clark 1958; Hendricks 1975; Davidson and Calpin 1976; Watkins and Nowell 1991). The remainder of the lifecycle occurs in blood-feeding H. reidi that ingests gamonts that then undergo syzygy, gamete maturation, syngamy, and sporogony in this definitive mite host. Large, polysporocystic oocysts typical of the genus Hepatozoon mature in the hemocoel of the mite definitive host and are presumably ingested by the squirrel host to initiate infection (Clark 1958; Redington and Jachowski 1971, 1972).
Infections with H. griseisciuri in eastern gray squirrels have been reported throughout the geographical range of the host, including the southeastern United States (Herman and Price 1955; Clark 1958; Parker 1968; Davidson and Calpin 1976), Rhode Island (Weidanz and Hyland 1958), and Wisconsin (Dorney and Todd 1959). The parasite has also been reported in the UK where the eastern gray squirrel has been introduced (Britt and Molyneux 1979; Watkins and Nowell 1991). It is unclear if these authors have all reported on the same Hepatozoon species. Perhaps several Hepatozoon species can utilize the eastern gray squirrel as an intermediate host. Classically, full characterization of a Hepatozoon sp. required detailed observations on parasite development in its definitive host to differentiate it from similar species. More recently, molecular data have been used to explore the diversity and relationships among Hepatozoon spp. and these data have revealed the existence of putative sibling species (e.g. Baneth et al. 2000). Currently, the molecular target from the adeleorinid coccidia with the most numerous publically available sequences is the nuclear 18S ribosomal RNA (rRNA) gene (rDNA). Extrachromosomal genome sequences from the mitochondrion and the apicoplast have become available recently from some members of the Adeleorina (Léveillé et al. 2014, 2019a, 2019b). There are currently no publicly available DNA sequences for H. griseisciuri.
We report the presence of intra-leukocytic gamonts consistent with the description of H. griseisciuri in Canadian eastern gray squirrels for the first time and describe the amplification and sequencing of 2 unique complete 18S rDNA sequences and 2 unique complete mitochondrial genome sequences from 3 infected eastern gray squirrels. These are the first DNA sequences reported for this parasite and allude to the presence of a pair of genetically distinct Hepatozoon species infecting Ontario eastern gray squirrels.
Materials and methods
Case material
Three adult road-killed eastern gray squirrels (S. carolinensis) were collected in Guelph, ON, Canada, necropsied and found to be infected with Hepatozoon cf. griseisciuri. Squirrel 1 was collected in June 2017; heart tissue containing coagulated blood was retained for DNA extraction. Squirrel 2 was collected in September 2017 and blood, liver, and spleen tissues were retained and frozen for DNA extraction. Squirrel 3 was collected in March 2018 and fresh samples of blood, liver, heart, kidney, spleen, and lung tissues were retained for histological observation and DNA extraction.
Histological slide preparation
During post-mortem examination, free-flowing blood was collected by syringe from the hearts of squirrel 2 and squirrel 3, and was used to make thin blood smears. Slides were air-dried, fixed, and stained with eosin and methylene blue (Shandon Kwik-Diff Stain, Thermo Fisher Scientific, Waltham MA, USA). Micrographs of gamont stages of Hepatozoon cf. griseisciuri were taken with a × 100 objective lens using bright-field illumination with an Olympus Provis AX70 microscope (Olympus, Tokyo, Japan) fitted with an Infinity 3-1C microscopy camera (Lumenera Corp., Ottawa, Canada).
Fresh liver, heart, kidney, spleen, and lung tissue collected from squirrel 3 were processed routinely after fixation in 10% buffered formalin for a 24–48 h period and were paraffin embedded. Briefly, 5-μm sections were deparaffinized with xylene and rehydrated by a series of graded baths in alcohol and phosphate-buffered saline buffer. All sections were stained with hematoxylin and eosin by the Animal Health Laboratory at the University of Guelph (Guelph ON, Canada).
DNA isolation
DNA was extracted from frozen blood-rich tissues (squirrel 1, heart; squirrel 2, lung; squirrel 3, liver) using a DNAzol reagent according to the manufacturer’s protocol (Thermo Fisher Scientific, Waltham MA, USA). After isolation, DNA was quantified spectrophotometrically using a Nanodrop 2000 instrument (Thermo Fisher Scientific, Waltham MA, USA).
Primer design
All existing and newly generated polymerase chain reaction (PCR) and sequencing primers (see Tables 1, 2, 3, and 4) were developed using the methods described in Léveillé et al. (2019b).
PCR amplification and amplicon isolation
High-fidelity PCR protocols and PCR amplicon isolation were carried out as described in Léveillé et al. (2019b). Complete nuclear 18S rDNA sequences were amplified from squirrel 1 (genotype A) and squirrel 3 (genotype B) samples as two overlapping amplicons generated using whole-gene universal primers Medlin_A and Medlin_B (forward and reverse primers, respectively, from Medlin et al., 1988) paired with Adeleorina-specific internal primers Adel_18S_1522R and Adel_18S_1000F respectively (Tables 1 and 2). A partial 18S rDNA sequence was amplified from squirrel 2 using the Medlin_A and Adel_18S_1522R primer pair.
The mitochondrial genome sequence of Hepatozoon cf. griseisciuri genotype A was generated from squirrel 1 from 4 overlapping amplicons. The first 2 amplicons were generated by a trial and error method that involved using all 4 combinations of forward and reverse primers targeting the highly conserved regions LSUF and LSUG (primers: Api_LSUF_F; Api_LSUF_R; Api_LSUG_F; Api_LSUG_R; primers cited in Table 1). Successful amplifications resulted in mitochondrial amplicon A1 generated from Api_LSUF_F and Api_LSUG_R and mitochondrial amplicon A2 generated from Api_LSUG_F with Api_LSUF_R (Table 1). After sequencing mitochondrial amplicon A1 and mitochondrial amplicon A2, primers Adel_RNA13_R and Hep_SSUB_F were selected to generate mitochondrial amplicon A3 that would span the LSUF region (Table 1). Primers Hep_CytB_391F and BarHep_COI_274R were selected to generate mitochondrial amplicon A4 to span the LSUG region (Table 1).
Initial attempts to amplify and sequence Hepatozoon cf. griseisciuri mitochondrial genome sequences from squirrel 2 yielded mixed sequences containing genotype A as well as a unique sequence, genotype B (results not shown). The new primer HepSq_B_RNA23t_F was designed to target genotype B specifically; this new primer was paired with Api_LSUG_R to generate mitochondrial amplicon B1 covering the majority of the mitochondrial genome sequence (Table 2). After sequencing mitochondrial amplicon B1, primer HepSq_B_COIR was designed to target genotype B specifically and paired with Hep_CytB_391F to generate mitochondrial amplicon B2 that overlapped both ends of mitochondrial amplicon B1 and completed the genotype B mitochondrial genome sequence (Table 2).
The mitochondrial genotype of squirrel 3 was identified by targeting a region spanning several fragmented rDNA that exhibited considerable variability between the H. griseisciuri genotype A and H. griseisciuri genotype B mitochondrial genomes (genotype A, 793 bp; genotype B, 747 bp). This region was targeted with universal primers Api_LSUE_F and Haem_RNA14_F (primers cited in Table 3). No additional sequencing of the mitochondrial genome from this isolate was done.
PCR amplicon sequencing and analysis
The purified PCR amplicons generated were then sequenced in both directions with the forward and reverse amplification primers supplemented with internal sequencing primers as needed (Tables 3 and 4), with exception of the partial 18S rDNA amplicon generated from squirrel 2. The latter amplicon was sequenced unidirectionally with the Medlin_A (F) primer (primer cited in Table 1). All Sanger sequencing was accomplished using an Applied Biosystems 3730 DNA Analyzer (Applied Biosystems Inc., Foster City CA, USA) by the Molecular Biology Unit of the Laboratory Services Division, University of Guelph (Guelph ON, Canada). Chromatograms received from sequencing reactions were imported and assembled into contigs using the De Novo Sequence Assembler within Geneious (version 6.1 or later, www.geneious.com, Kearse et al. 2012). All assembled amplicon contigs overlapped one another by at least 250 bp and overlapping regions showed 100% pairwise sequence identity in all cases.
Complete 18S rDNA sequences from genotype A and genotype B were searched against the public sequence databases using the BLAST algorithm (Altschul et al. 1990) to identify similar 18S rDNA sequences.
Annotation and analysis of protein-coding regions
Open reading frames (ORF) in the assembled mitochondrial genome were identified using an ORF search utility within Geneious using transl_table 4 (i.e., mold protozoan mitochondrial translation table). Translated ORFs (amino acid residue (AA) sequences) were searched against the public sequence databases using the DELTA-BLAST algorithm (Altschul et al. 1990; Boratyn et al. 2012) to identify each ORF in relation to similar protein-coding sequences (CDS).
Annotation of mitochondrial ribosomal DNA sequences
The fragmented mitochondrial rDNA of H. griseisciuri genotype A and genotype B were annotated based on similarity to the fragmented rDNA previously annotated in Plasmodium falciparum (M76611; Feagin et al. 2012), Hepatozoon catesbianae (KF894962; Léveillé et al. 2014), Klossiella equi (MH203050; Léveillé et al. 2019b), and Eimeria tenella (HQ702484) mitochondrial genomes as described by Léveillé et al. (2014, 2019a, 2019b). Any rDNA that aligned with the H. griseisciuri mitochondrial sequence with a pairwise sequence identity below 65% were excluded from further analysis. Putative fragmented rDNA ribosomal annotations (Table 4) follow the nomenclature of Feagin et al. (2012) in their annotation of the mitochondrial genome of P. falciparum.
Pairwise comparison of Hepatozoon cf. griseisciuri genotype A and genotype B
Pairwise comparisons of 18S rDNA, whole mitochondrial genome sequences and orthologous CDS were analyzed in Geneious using the “Multiple Align” tool (cost matrix: 70%, gap open penalty: 12, gap extension penalty: 3). Pairwise comparisons of translated AA sequences from orthologous CDS were analyzed in Geneious using the “Multiple Align” tool (cost matrix: Blosum62, gap open penalty: 12, gap extension penalty: 3).
Results
Microscopic observations
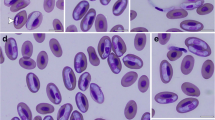
Blood smears were not available for Squirrel 1 and therefore microscopic observations of gamonts were not possible. Stained thin blood smears collected from Squirrel 2 and Squirrel 3 contained infrequent Hepatozoon cf. griseisciuri gamonts found within blood cells that most resembled monocytes (Fig. 1). Gamonts observed from Squirrel 2 (n = 8) measured 10.4–11.4 (10.9) μm × 3.5–5.0 (4.1) μm with gamont nuclei measuring 4.2–6.9 (5.4) μm × 2.7–4.1 (3.1) μm. Gamonts observed from Squirrel 3 (n = 9) measured 9.3–11.3 (10.6) μm × 3.2–5.0 (3.9) μm with gamont nuclei measuring 3.9–7.0 (5.0) μm × 2.5–3.6 (3.1) μm.

Micrographs of Giemsa’s stained gamonts of Hepatozoon cf. griseisciuri in a thin blood film from eastern gray squirrel, Sciurus carolinensis, located within a monocyte (a) and free (b) in the blood. Scale bar = 10 μm
There were no parasite stages observed from the histological sections of liver, heart, kidney, spleen, and lung tissues recovered from Squirrel 3.
Complete nuclear 18S ribosomal DNA sequences
The complete nuclear 18S rDNA sequence of H. griseisciuri genotype A amplified from squirrel 1 had a unit length of 1816 bp (MK452252). The complete nuclear 18S rDNA genotype B amplified from squirrel 3 also had a unit length of 1816 bp (MK452253). A pairwise comparison of the 2 sequences had a 98.8% pairwise identity and 22 single-nucleotide differences (SND). Both sequences generated similar BLAST search results with 98–99% pairwise identity (92–98% coverage) to several unnamed Hepatozoon sp. 18S rDNA sequences (AB181504, AY600626, FJ719818, FJ719816, JX644997, EF222259) collected from various rodent hosts and 99% pairwise identity (97% coverage) to a Hepatozoon ayorgbor 18S rDNA sequence (EF157822) collected from a ball python (Python regius).
The partial 18S rDNA amplicon generated from squirrel 2 (829 bp; sequence not published) had 19 ambiguous base calls (caused by multiple base signals contributing to the chromatogram at these sites). The 19 ambiguous base calls documented in the squirrel 2 partial 18S rDNA sequence corresponded to the SNDs observed between genotype A and genotype B 18S rDNA sequences. This confirmed that squirrel 2 was infected with both genotype A and genotype B of H. griseisciuri with no other genotypes detected.
Complete mitochondrial genome sequences
The complete mitochondrial genome of H. griseisciuri genotype A amplified from squirrel 1 had a unit length of 6311 bp with an overall G+C content of 39.2% (MK452388). The circular-mapping mitochondrial genome organization (Fig. 2) consisted of three protein-coding genes cytochrome c oxidase subunit I (COI), cytochrome c oxidase subunit III (COIII), and cytochrome b (CytB) (Table 5). The G+C content of these protein-coding genes was 37.6% for COI, 36.4% for COIII, and 37.0% for CytB. The complete mitochondrial genome of H. griseisciuri genotype B selectively amplified from squirrel 2 had a unit length of 6114 bp with an overall G+C content of 38.6% (MK452389). The mitochondrial genome organization was nearly identical to that of genotype A and also contained three protein-coding genes (COI, COIII, and CytB); the G+C content of these protein-coding genes was 37.1% for COI, 34.4% for COIII, and 36.6% for CytB. As is typical of apicomplexan mitochondrial genomes, many putative fragmented rDNA were identified in both genome sequences: 14 fragmented large subunit (LSU) rDNA; 10 fragmented small subunit (SSU) rDNA; and 8 unassigned fragmented rDNA (Table 6). Each putative fragmented rDNA had a pairwise identity greater than 65% to a corresponding fragmented rDNA functionally annotated previously in the mitochondrial genome of P. falciparum (M76611; see Feagin et al. 2012). As observed in the mitochondrial genome of K. equi (Léveillé et al. 2019b), the sequence for LSUA was fragmented in 2 pieces with the start and the end of the gene occurring at different locations. The sequence identified as RNA16 overlapped with the 3′-end of the COI CDS in both genomes. Fragmented rDNA encoding RNA8, RNA11, and RNA12 in the P. falciparum mitochondrial genome were not identified in either of the mitochondrial genotypes from Hepatozoon cf. griseisciuri (Table 6).

Annotated complete mitochondrial genome sequences of Hepatozoon cf. griseisciuri genotype A (MK452388), illustrated in both circular and linear forms, and genotype B (MK452389), illustrated in linear form only. Three protein-coding regions (CDS; cytochrome c oxidase subunit I (COI), cytochrome c oxidase subunit III (COIII), and cytochrome b (CytB)) were encoded by the genotype A and genotype B mitochondrial genomes. Numerous putative fragmented ribosomal RNA (rRNA) genes (rDNA) were identified in both genomes; fragmented rDNA nomenclature follows Feagin et al. (2012). The arrangements of CDS and fragmented rDNA were identical in the genotype A and genotype B mitochondrial genomes with the exception of RNA2 that was found in different locations on each (*). Initial genome images were produced with Geneious (www.geneious.com; Kearse et al. 2012)
The small (747 bp) mitochondrial genome target amplified from squirrel 3 using primers that target both genotypes had 100% pairwise identity to the genotype B sequence selectively amplified from squirrel 2. This sequence contained no ambiguous base calls (caused by competing chromatogram readings) indicating that the squirrel 3 sample contained genotype B exclusively.
Although genotype A and genotype B mitochondrial genomes had the same CDS and rDNA elements, they differed slightly in their organization. RNA2 mapped near the start of CytB in genotype A but was located upstream of COIII and reading in the opposite direction in genotype B (Fig. 2, Table 6). There was an 8-bp insert (TAAGTAAG; location: 1418–1425 bp) near the end of the genotype B COI CDS that resulted in a frameshift mutation and caused the COI CDS to be longer than the genotype A COI CDS (Table 5). The genotype B COI CDS had a 27-bp overlap with the end of the COIII CDS. There was also a 26-bp insert near the end of the genotype A CytB CDS that terminated the coding region earlier than the genotype B CytB CDS (Table 5). Pairwise alignment of genotype A and genotype B complete mitochondrial genome sequences had a pairwise identity of 89.1%. Pairwise identity values of protein CDS and translation alignments are shown in Table 5.
When searched against public databases using the DELTA-BLAST algorithm (Altschul et al. 1990; Boratyn et al. 2012), the translation predictions of mitochondrial protein-coding sequences from H. griseisciuri genotype A and genotype B were found to be similar to the few currently published adeleorinid and various hemosporinid mitochondrial genome sequences (see Table 7).
Discussion
We report the first molecular data (complete nuclear 18S rDNA and the complete mitochondrial genome) from Hepatozoon spp. that is consistent with the only Hepatozoon species described to infect the eastern gray squirrel, Hepatozoon griseisciuri Clark, 1958. The pairwise differences observed between 18S rDNA and mitochondrial genome sequences of genotype A and genotype B indicate that these genotypes likely represent two different Hepatozoon spp. infecting eastern gray squirrels in Ontario. The 0.08 divergence at the COI locus versus the 0.012 divergence between 18S rDNA sequences observed in the present study supports the use of the former genetic locus for species-level analyses, particularly given the proclivity of 18S rDNA in the nuclear genomes of apicomplexan parasites to have multiple paralogous copies (see Léveillé et al. 2019a). The superiority of COI sequences over nuclear 18S rDNA for species delimitation has been demonstrated previously for eimeriid coccidia (Ogedengbe et al. 2011); drawing the same conclusion for adeleorinid coccidia will require additional sequence data from the mitochondrial genomes of these parasites. Pending further investigations on the biology and lifecycles of these parasites, we have taken the position that the two genotypes described herein should both retain the name of Hepatozoon cf. griseisciuri. Only after their biologies can be linked unequivocally with a specific genotype can one, both, or neither genotype be associated unequivocally with H. griseisciuri. The presence of both H. griseisciuri genotypes infecting squirrel 2 reveal that these related species are capable of infecting the same intermediate host concurrently.
The morphometrics of gamonts within monocytes from squirrel 2 (genotype A and genotype B) and squirrel 3 (genotype B) were not significantly different from one another (data not shown) and both corresponded to the dimensions (9.9–11.8 × 3.3–4 μm; nuclei, 4–5.2 × 3.3 μm (n = 5)) provided by Clark (1958) in his description of H. griseisciuri. No merogonic stages were observed histologically in the tissues collected from squirrel 3.
Searching the 18S rDNA sequences of H. griseisciuri genotype A and genotype B against public databases using the BLAST algorithm (Altschul et al. 1990) confirmed that there are several unnamed Hepatozoon spp. sampled from various rodent intermediate hosts worldwide with high pairwise sequence identities (98–99%). Surprisingly, the 18S rDNA sequences from H. griseisciuri also had high pairwise sequence identity (99%) to an 18S rDNA sequence from H. ayorgbor infecting a ball python. Hepatozoon ayorgbor was demonstrated experimentally to have a 3-host lifecycle involving a mosquito definitive host, a rodent first intermediate host, and a serpent second intermediate host (Sloboda et al. 2007, 2008).
The complete mitochondrial genomes of the H. griseisciuri (genotype A and genotype B) infecting the eastern gray squirrel were structurally similar to the majority of described mitochondrial genomes from the Apicomplexa: circular mapping; 3 CDS (COI, COIII, CytB); and multiple fragmented small and large subunit rDNAs (e.g., Hikosaka et al. 2011a, 2013; Feagin et al. 2012; Léveillé et al. 2014, 2019b). As with many apicomplexan mitochondrial genomes, the physical form of these circular-mapping genomes of these H. griseisciuri genotypes could be either linear concatemers of several to many genome copies or single physically circular genomes (see Hikosaka et al. 2013).
Beyond basic gene content comprising each of the mitochondrial genomes, the organization of those genes in the H. griseisciuri (genotype A and genotype B) mitochondrial genomes was unique among the Apicomplexa, including mitochondrial genomes from other adeleorinid coccidia (Léveillé et al. 2014, 2019a, b) and, surprisingly, the two genotypes sequenced in the present study had minor differences in organization. This is in contrast to the usual conservation of mitochondrial genome organization (i.e., the order and orientation of these genome components) observed for members within each of several major apicomplexan groups (e.g., the hemosporinids (see Hikosaka et al. 2011b) or eimeriids) but not between major groups. Among eimeriid coccidia, the mitochondrial genome organization and content is virtually identical among 5 different genera (i.e., the genera Eimeria, Isospora, Caryospora, Cyclospora, and Lankesterella (see Ogedengbe 2015)); the degree of reorganization observed between the two Hepatozoon genotypes in the present paper exceeded the variation reported among these eimeriid parasites. Among other sequenced apicomplexan mitochondrial genomes, only the piroplasms (Babesia spp. and Theileria spp.) seem to display a diversity of mitochondrial genome structures within single genera (Hikosaka et al. 2010).
Despite the observed organizational diversity, the mitochondrial genomes of Hepatozoon cf. griseisciuri did share similarities with those of H. catesbianae and K. equi. The mitochondrial genome of genotype A had a 76-bp repeat region from 21-bp upstream to 55 bp into the CDS of both COIII and CytB, and the genotype B mitochondrial genome had an 85 bp from 30-bp upstream to 55 bp into the CDS of both COIII and CytB. Similar repeated regions were observed at the starts of the COIII and CytB CDS of H. catesbianae (70 bp; see Léveillé et al. 2014) and K. equi (107 bp; see Léveillé et al. 2019b). In all cases, these repeated stretches did not seem to alter the predicted transmembrane folding structures of the resulting translated AA sequences (not shown). There was also a common cluster of fragmented rDNA that were conserved among the genomes. A cluster of fragmented rDNA (LSUD-RNA23t-RNA1-RNA22-LSUG) was conserved in H. griseisciuri, H. catesbianae, and K. equi mitochondrial genomes. Further, the mitochondrial genome of H. catesbianae shared several long stretches of fragmented rDNA (LSUC-LSUF-RNA18-SSUF; RNA6-RNA14-SSUB-RNA15-LSUA(2)-RNA7; RNA3-SSUD-RNA10-LSUE; RNA20-RNA9-RNA4-LSUA(1)-CytB) with the two H. griseisciuri genotypes. Some of these elements (SSUF, LSUF, LSUC, RNA14, SSUB) clustered closely together in the mitochondrial genome of K. equi, but were not in the same arrangement. This suggests that fewer rearrangement events have occurred within the mitochondrial genomes of H. griseisciuri and H. catesbianae compared with the mitochondrial genome of K. equi. These similarities and the higher pairwise identities of the nuclear 18S rDNA and mitochondrial CDS sequences of H. catesbianae and H. griseisciuri compared with K. equi reinforces that these Hepatozoon spp. are more closely related to one another than to K. equi.
The mitochondrial genomes of Hepatozoon spp. sequenced to date display staggering diversity (see Chapter 8 of Léveillé 2019). This is reinforced by the apparent absence of a complete mitochondrial genome sequence in Hepatozoon canis (see Léveillé et al. 2019a). While we have only begun to scratch the surface of this diversity, observations to date suggest that taxa currently in the genus Hepatozoon are likely distributed into multiple genera that have yet to be defined. The likely necessity to split the genus Hepatozoon has been recognized previously (see Smith and Desser 1997). Karadjian et al. (2015) started the required taxonomic revisions based on a combined analysis of the biology of multiple hemogregarines (sensu lato) and a large-scale phylogenetic analysis of adeleorinid 18S rDNA sequences. As a result, Karadjian et al. (2015) described a new genus, Bartazoon, to hold all members of the “hemogregarines type 4” clade in their phylogenetic tree. Our data suggest that their proposed taxonomic solution does not sufficiently address the breadth of diversity observed among these heteroxenous adeleorinid parasites; the number of genera may need to be increased to accommodate this diversity.
Our preliminary analysis of the new 18S rDNA sequences obtained herein suggests that H. griseisciuri (genotype A and genotype B) would also cluster within the “hemogregarines type 4” clade of Karadjian et al. (2015) (data not shown, see Figure 8.2 from Léveillé 2019). The biology of H. griseisciuri involves a rodent intermediate host and mite definitive host with transmission presumably via ingestion of mites containing sporulated oocysts (Clark 1958; Redington and Jachowski 1971). Although Hepatozoon species have widely varying lifecycles in a range of biologically diverse definitive and intermediate hosts (Smith 1996; Karadjian et al. 2015), the biology of H. griseisciuri (see Clark 1958) is most similar to the Hepatozoon species used to define the genus Hepatozoon by Miller (1908), with which it shares both geography (eastern North America) and a definitive acarine host, at least experimentally (Clark 1958). If the adeleorinid nuclear and mitochondrial genotypes explored in the present paper are truly congenic with the type species of the genus Hepatozoon as described by Miller (1908); then, the assignment of all species that clustered in “hemogregarines type 4” to the genus Bartazoon by Karadjian et al. (2015) becomes problematic. This was supported by the conclusions of Maia et al. (2016) that reinforced the need for a systematic revision of the Adeleorina but warned that it would be preferable to wait to name distinct lineages formally until additional molecular data became available, particularly sequences from the currently accepted type species, H. muris.
The amplification and sequencing of partial 18S rDNA and complete mitochondrial genome sequences from Hepatozoon cf. griseisciuri uncovered two, genotypically distinct Hepatozoon species found infecting eastern gray squirrels both singly and concurrently; this unexpected genetic diversity foreshadows the diversity among adeleorinid blood parasites that remains to be uncovered. This reinforces that the taxonomic revision of this group continues to be complicated by the comparable lack of molecular data linked to well-defined biological characteristics (Smith 1996). The majority of adeleorinid species have only ever been observed in one (of perhaps several) intermediate host and, frequently, solely from the easily observable stage in the lifecycle, the intracellular gamont. Moreover, the definitive hosts, within which the majority of the biologically distinctive lifecycle stages (gametogonic and sporogonic) are found, can be challenging to collect and are labor intensive to study in the laboratory. As sequences from multiple genetic loci from adeleorinid parasites become increasingly available, these molecular markers can be applied to link morphological and biological observations among the various hosts and tissues infected during these complex heteroxenous lifecycles. Only then will we be able to begin to understand the biology and evolutionary relationships among these diverse adeleorinid taxa.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Baneth G, Barta JR, Shkap V, Martin DS, Macintire DK, Vincent-Johnson N (2000) Genetic and antigenic evidence supports the separation of Hepatozoon canis and Hepatozoon americanum at the species level. J Clin Microbiol 38:1298–1301
Barta JR (2000) Suborder Adeleorina Leger, 1911. In: Lee JJ, Leedale GF, Bradbury P (eds) An illustrated guide to the Protozoa, 2nd edn. Society of Parasitologists, Lawrence, Kansas, pp 305–318
Boratyn GM, Schäffer AA, Agarwala R, Altschul SF, Lipman DJ, Madden TL (2012) Domain enhanced lookup time accelerated BLAST. Biol Direct 7:1–15. https://doi.org/10.1186/1745-6150-7-12
Britt D, Molyneux DH (1979) Parasites of grey squirrels in Cheshire, England. J Parasitol 65:408. https://doi.org/10.2307/3280284
Clark GM (1958) Hepatozoon griseisciuri n. sp.; a new species of Hepatozoon from the grey squirrel (Sciurus carolinensis Gmelin, 1788), with studies on the lifecycle. J Parasitol 44:52–63. https://doi.org/10.2307/3274829
Coles AC (1914) Blood parasites found in mammals, birds and fishes in England. Parasitology 7:17–61. https://doi.org/10.1017/S0031182000006284
Davidson WR, Calpin JP (1976) Hepatozoon griseisciuri infection in gray squirrels of the southeastern United States. J Wildl Dis 12:72–76. https://doi.org/10.7589/0090-3558-12.1.72
Dorney RS, Todd AC (1959) Occurrence of Hepatozoon in Gray Squirrels in Wisconsin. J Parasitol 45:309. https://doi.org/10.2307/3274506
Feagin JE, Harrell MI, Lee JC, Coe KJ, Sands BH, Cannone JJ, Tami G, Schnare MN, Gutell RR (2012) The fragmented mitochondrial ribosomal RNAs of Plasmodium falciparum. PLoS One 7:e38320. https://doi.org/10.1371/journal.pone.0038320
Hendricks LD (1975) Schizogonic development of Hepatozoon griseisciuri Clark 1958 (Sporozoa: Haemogregarinidae), of the gray squirrel (Sciurus carolinensis Gmelin 1788). J Parasitol 61:458–461. https://doi.org/10.2307/3279324
Herman CM, Price DL (1955) The occurrence of Hepatozoon in the gray squirrel (Sciurus carolinensis). J Protozool 2:48–51. https://doi.org/10.1111/j.1550-7408.1955.tb02396.x
Hikosaka K, Kita K, Tanabe K (2013) Diversity of mitochondrial genome structure in the phylum Apicomplexa. Mol Biochem Parasitol 188:26–33. https://doi.org/10.1016/j.molbiopara.2013.02.006
Hikosaka K, Nakai Y, Watanabe Y, Tachibana S-I, Arisue N, Palacpac NMQ, Toyama T, Honma H, Horii T, Kita K, Tanabe K (2011a) Concatenated mitochondrial DNA of the coccidian parasite Eimeria tenella. Mitochondrion 11:273–278. https://doi.org/10.1016/j.mito.2010.10.003
Hikosaka K, Watanabe Y-I, Kobayashi F, Waki S, Kita K, Tanabe K (2011b) Highly conserved gene arrangement of the mitochondrial genomes of 23 Plasmodium species. Parasitol Int 60:175–180. https://doi.org/10.1016/j.parint.2011.02.001
Hikosaka K, Watanabe YI, Tsuji N, Kita K, Kishine H, Arisue N, Palacpac NMQ, Kawazu SI, Sawai H, Horii T, Igarashi I, Tanabe K (2010) Divergence of the mitochondrial genome structure in the apicomplexan parasites, Babesia and Theileria. Mol Biol Evol 27:1107–1116. https://doi.org/10.1093/molbev/msp320
Karadjian G, Chavatte J-M, Landau I (2015) Systematic revision of the adeleid haemogregarines, with creation of Bartazoon n. g., reassignment of Hepatozoon argantis Garnham, 1954 to Hemolivia , and molecular data on Hemolivia stellata. Parasite 22:31. https://doi.org/10.1051/parasite/2015031
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Léveillé AN (2019) Scratching the surface: Diversity among the first sequenced extrachromosomal genomes of parasites in the suborder Adeleorina (Apicomplexa) with a focus on Hepatozoon species. Doctor of Philosophy Dissertation, University of Guelph. http://hdl.handle.net/10214/16973
Léveillé AN, Baneth G, Barta JR (2019a) Next generation sequencing from Hepatozoon canis (Apicomplexa: Coccidia: Adeleorina): complete apicoplast genome and multiple mitochondrion-associated sequences. Int J Parasitol 49:375–387. https://doi.org/10.1016/j.ijpara.2018.12.001
Léveillé AN, Bland SK, Carlton K, Larouche CB, Kenney DG, Brouwer ER, Lillie BN, Barta JR (2019b) Klossiella equi infecting kidneys of Ontario horses: lifecycle features and multi locus sequence based genotyping confirm the genus Klossiella belongs in the Adeleorina (Apicomplexa: Coccidia). J Parasitol 105:29–41. https://doi.org/10.1645/18-80
Léveillé AN, Ogedengbe ME, Hafeez MA, Tu H-HA, Barta JR (2014) The complete mitochondrial genome sequence of Hepatozoon catesbianae (Apicomplexa: Coccidia: Adeleorina), a blood parasite of the green frog, Lithobates (formerly Rana) clamitans. J Parasitol 100:651–656. https://doi.org/10.1645/13-449.1
Maia JP, Carranza S, Harris DJ (2016) Comments on the systematic revision of adeleid haemogregarines: are more data needed? J Parasitol 102:549–552. https://doi.org/10.1645/15-930
Medlin L, Elwood HJ, Stickel S, Sogin ML (1988) The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 71:491–499. https://doi.org/10.1016/0378-1119(88)90066-2
Miller WW (1908) Hepatozoon perniciosum (n.g., n.sp.) : a haemogregarine pathogenic for white rats, with a description of the sexual cycle in the intermediate host, a mite (Lelaps echidninus). Washington: Government Printing Office
Ogedengbe ME (2015) DNA barcoding of Apicomplexa: mitochondrial evolution across the phylum. Doctor of Philosophy Dissertation, University of Guelph
Ogedengbe JD, Hanner RH, Barta JR (2011) DNA barcoding identifies Eimeria species and contributes to the phylogenetics of coccidian parasites (Eimeriorina, Apicomplexa, Alveolata). Int J Parasitol 41:843–850. https://doi.org/10.1016/j.ijpara.2011.03.007
Parker JC (1968) Parasites of the gray squirrel in Virginia. J Parasitol 54:633. https://doi.org/10.2307/3277102
Redington BC (1970) Studies on the morphology and taxonomy of Haemogamasus reidi Ewing, 1925 (Acari: Mesostigmata). Acarologia 12:643–667
Redington BC, Jachowski LA (1971) Syngamy and sporogony of Hepatozoon griseisciuri Clark, 1958 (Sporozoa: Haemogregarinidae), in its natural vector, Haemogamasus reidi Ewing, 1925 (Acari: Mesostigmata). J Parasitol 57:953–960. https://doi.org/10.2307/3277842
Redington BC, Jachowski LA (1972) Role of Haemogamasus reidi (Acari: Mesostigmata) in the lifecycle of the gray squirrel protozoan, Hepatozoon griseisciuri (Sporozoa: Haemogregarinidae). J Parasitol 58:401–403. https://doi.org/10.2307/3278114
Sloboda M, Kamler M, Bulantová J, Votýpka J, Modrý D (2007) A new species of Hepatozoon (Apicomplexa: Adeleorina) from Python regius (Serpentes: Pythonidae) and its experimental transmission by a mosquito vector. J Parasitol 93:1189–1198. https://doi.org/10.1645/GE-1200R.1
Sloboda M, Kamler M, Bulantová J, Votýpka J, Modrý D (2008) Rodents as intermediate hosts of Hepatozoon ayorgbor (Apicomplexa: Adeleina: Hepatozoidae) from the African ball python, Python regius? Folia Parasitol (Praha) 55:13–16. https://doi.org/10.14411/fp.2008.003
Smith TG (1996) The genus Hepatozoon (Apicomplexa: Adeleina). J Parasitol 82:565–585. https://doi.org/10.2307/3283781
Watkins BM, Nowell F (1991) Hepatozoon in gray squirrels (Sciurus carolinensis) trapped near Reading, Berkshire. J Zool 224:101–112. https://doi.org/10.1111/j.1469-7998.1991.tb04791.x
Weidanz WP, Hyland KE (1958) The occurrence of Hepatozoon sciuri in gray squirrels in New England. J Parasitol 44:97. https://doi.org/10.2307/3274836
Acknowledgments
A.N.L. was supported by a Queen Elizabeth II Graduate Scholarship in Science and Technology and an Ontario Graduate Scholarship from the Government of Ontario, and an OVC Scholarship from the Ontario Veterinary College, University of Guelph. Dr. Darren Wood (Pathobiology, University of Guelph) is thanked for his assistance in the specific identification of parasitized squirrel leukocytes. Dr. Todd Smith, Perryn Kruth, Rachel Imai, and Jessica Rotolo are thanked for their comments on earlier drafts of this paper; Perryn Kruth is also thanked for her assistance with squirrel necropsies. The staff of the Guelph Molecular Supercenter, Laboratory Services Division, University of Guelph, are thanked for their diligent hard work.
Funding
This research was supported by a Discovery Grant to J.R.B. from the Natural Sciences and Engineering Research Council of Canada (NSERC-DG #400566).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Section Editor: Domenico Otranto
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Léveillé, A.N., El Skhawy, N. & Barta, J.R. Multilocus sequencing of Hepatozoon cf. griseisciuri infections in Ontario eastern gray squirrels (Sciurus carolinensis) uncovers two genotypically distinct sympatric parasite species. Parasitol Res 119, 713–724 (2020). https://doi.org/10.1007/s00436-019-06583-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-019-06583-5