
Abstract
Standard diagnostic methods currently in use for the identification of helminth infections in ruminants are based on the morphological analysis of immature and adult stages of parasites. This paper describes a method for the semiquantitative identification of nematodes, mainly Trichostrongyloidea, at species-level resolution. The method is based on amplification and fragment analysis followed by minisequencing of the ITS-2 region (internal transcribed spacer 2) of the ribosomal DNA of parasite eggs or larvae. This method allows for the identification of seven genera (Chabertia, Cooperia, Haemonchus, Oesophagostomum, Ostertagia, Teladorsagia, and Trichostrongylus) and 12 species (Chabertia ovina, Cooperia curticei, Cooperia punctata, Cooperia oncophora/Cooperia surnabada, Haemonchus contortus, Haemonchus placei, Haemonchus longistipes, Oesophagostomum asperum, Oesophagostomum radiatum, Ostertagia ostertagi, Trichostrongylus axei, and Trichostrongylus colubriformis) of infectious nematodes of domestic ruminants. The concordance between the morphological and molecular analyses in the detection of genera ranged from 0.84 to 0.99, suggesting the proposed detection method is specific, semiquantitative, less laborious, and highly cost-efficient.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Domestic ruminants are infected by various species of gastrointestinal nematodes, whose proportion in the host may vary according to individual susceptibility, environmental contamination, rainy or dry season, and sensitivity of the species to anthelmintic drugs (Dobson et al. 1996). Numerous reports have described resistance to different classes of anthelmintics in sheep (Torres-Acosta et al. 2012; Salgado and Santos 2016), cattle (Sutherland and Leathwick 2011), and goats (Várady et al. 2007; Coelho et al. 2010), making it increasingly prevalent.
Given the wide diversity of infectious parasite species and the growing problem of anthelmintic resistance (Kaplan 2004), it is essential to gain a deeper understanding of several epidemiological aspects in order to face the current challenges in the control of gastrointestinal nematodes (Charlier et al. 2018). Parasite populations in herds are not static but vary according to climate, management strategies, and the transportation of animals (Hansen and Perry 1994); hence, prevalence is an important measure of risk factors for herd health (Raue et al. 2017). Moreover, the same parasitic load of different species may have different pathogenic potentials (Ueno and Gonçalves 1998). Thus, the precise identification of different species, as well as knowledge about the epidemiology of parasitic infections, is fundamental for the development of sustainable parasite prevention and control strategies.
The main means of diagnosing nematodes in ruminants is through coproparasitological tests, whereby nematode genera are identified based on an analysis of eggs and infective larvae found in feces. Egg from Strongyloides spp., Trichuris spp., and Toxocara vitulorum easily can be identified based on shape and size. A “strongyle” egg is not often possible to identify to genus level as the eggs of most species are similar in appearance and overlapping in size. Exception are Nematodirus spp. eggs which are larger than the typical egg of a “strongyle” egg (i.e., Haemonchus, Trichostrongylus, Cooperia, Ostertagia, Bunostomum, Chabertia, etc.). Thus, fecal culture and morphometric analysis of third-stage larvae (L3) must be done to identify the genera that are present (Van Wyk et al. 2004). Except for the identification of some eggs, these tests are not only difficult (Bott et al. 2009) but also time-consuming and requiring training to identify the morphology of the parasitic stage (Roeber et al. 2017a). To circumvent this situation, molecular diagnostic tools have been proposed to assist in the diagnosis of parasitic species, in the monitoring of parasite prevalence and distribution, and in the detection of anthelmintic resistance (Roeber et al. 2017b). Additionally, the PCR-based methods have increased in popularity in the scientific community (Amarante et al. 2017; Rashid et al. 2018). More recently, nemabiome approaches through deep sequencing have been used to estimate the relative abundance of different species of gastrointestinal strongylid of ruminants (Avramenko et al. 2015, 2017, 2018; Redman et al. 2019).
Methods based on the detection of DNA, which can be extracted from specimens at any stage of development, have advantages over traditional microscopy techniques because they do not require the culturing of nematodes. They are more sensitive and specific for quantification and can be used cost effectively on a large scale through automated testing platforms (Gilleard 2006; Von Samson-Himmelstjerna 2006; Roeber et al. 2011). Moreover, molecular methods can potentially help shed light on cross-infection between ruminants because they are more accurate at the species level, and they are important tools in the diagnosis of anthelmintic resistance (Ljungström et al. 2018).
Among these methods, several molecular assays have been proposed for the identification or differentiation of strongyle species using genetic markers such as ribosomal or mitochondrial spacer regions (Gasser 2006). The regions explored for this purpose have low intraspecific variability and high interspecific variability (Gasser et al. 1997), which justifies their use as molecular tools for species identification and quantification (Nielsen et al. 2008; Learmount et al. 2009; Roeber et al. 2012). However, the joint and simultaneous evaluation of multiple nematode species is hindered by the fact that techniques based on conventional or real-time PCR lose reproducibility and accuracy as the number of primers in the reaction mix (multiplex) increases, particularly due to the multiple possible interactions between them during the amplification of target DNA (Zarlenga et al. 2001; Höglund et al. 2013; Roeber et al. 2017a, b). Hence, there is urgent need for the development of new methods for the identification of nematode parasites of ruminants. A possible alternative is based on the presence of insertion-deletion polymorphisms (indels) in specific sequences. The internal transcribed spacer (ITS) regions of ribosomal DNA generally contain many indel polymorphisms, and interspecific differences in these polymorphisms are a particularly useful characteristic for taxonomic identification (Blouin 2002). Besides, this region has conserved sequences between different genera, which allows an analysis of the fragment using only one primer pair.
This paper describes the development of a process based on the use of a universal ITS-2 region primer pairs for the semiquantitative identification of the main gastrointestinal nematodes of domestic ruminants contained in samples of eggs, larvae, or adult worms through of PCR followed by capillary electrophoresis and PCR followed by minisequencing.
Material and methods
Standardization, development, and demonstration of the efficacy of the method for the molecular identification of gastrointestinal strongyles in domestic ruminants were done.
Samples
The technique was developed, standardized, and validated using samples of eggs, L3 larvae, and adults of gastrointestinal nematodes collected from cattle, and in some cases, also from goats, sheep, and buffaloes. The source of the samples and the methodology used to obtain them are described below.
Eggs
Fecal samples from cows (n = 100) on a farm located in Uberaba, state of Minas Gerais (MG) were extracted directly from the animals’ rectums into plastic bags, which were then labeled and stored at 4 °C. The number of eggs per gram (EPG) of feces in the samples was calculated following the technique described by Gordon and Withlock (1939). After the EPG count, eggs diluted in TEN buffer solution (1 M Tris-HCl, 0.5 M EDTA, 5 M NaCl) in a Petri dish were examined under an optical microscope, collected individually with micropipette, and placed separately in microtubes containing 5 μl of TEN buffer, which were stored at 4 °C.
Larvae
L3 larvae were obtained from pools (n = 171) of fecal samples collected from cows and sheep on farms located in the states of Minas Gerais and Rio de Janeiro, respectively. The samples that presented the highest EPG counts were subjected to coproculture according to the methodology described by Roberts and O'Sullivan (1950), after which the Baermann technique (Cort et al. 1922) was used to recover the L3 larvae. The sediment containing the L3 was resuspended in absolute ethanol, and then placed for 4 min on a Petri dish containing 2.5% sodium hypochlorite solution. The L3 larvae were then collected as previously mentioned to eggs and stored individually at 4 °C in TEN buffer.
In preparation for the morphometric identification of L3, the samples were centrifuged at 2000×g for 1 min. They were then divided into aliquot parts, which were mounted on microscope slides, stained with Lugol’s iodine at 5%, and then identified (100 larvae per sample) to determine the percentage of genera. The nematode genera were identified using the methodology proposed by Van Wyk et al. (2004) and Van Wyk and Mayhew (2013).
Adults
Adult worm specimens were obtained from cattle, sheep, and goats of the collection used by Brasil et al. (2012).
Method for the molecular identification of the main species and genera of gastrointestinal nematodes of domestic ruminants
The gastrointestinal nematode species were subjected to molecular identification in four main steps basically involving the following: (i) extraction of DNA from the nematode specimen to be analyzed; (ii) preliminary PCR for partial amplification of the ITS-2 region using universal nematode primers; (iii) minisequencing-based analysis of fragments to identify the genera Haemonchus, Cooperia, Oesophagostomum, Trichostrongylus, Ostertagia, Teladorsagia, and Chabertia; and (iv) examination of the species of the genera identified by means of specific primers for identification by DNA minisequencing.
Extraction of DNA from samples (step 1)
For individual analysis of specimens, DNA was extracted by adding 5 μl of extraction buffer (1 mM Tris-HCl, 0.1 mM EDTA, and 5 mg/ml proteinase K) to the individual egg or L3 specimens. To extract DNA from adult worms, 50 μl of the same extraction buffer was added. The samples were then allowed to be stored overnight at 41 °C. After this procedure, they were subjected to a temperature of 95 °C for 20 min and then stored at − 20 °C until the moment the molecular analysis was performed (adapted from Silvestre and Humbert 2000). For the analysis of a pool of specimens, 10 μl of 2.5% sodium hypochlorite solution, followed by 20 μl of 0.8% NaOH solution, were added to samples consisting of a pool of eggs or L3 larvae.
The DNA extraction was successful for both egg pool and larvae. Eggs (n = 100) and larvae (n = 100) of the same sample were separated individually and the fragment analysis was performed. Both analyses showed similar proportions of genera (data not shown).
Samples were kept in a bath at 97 °C for 15 min, followed by the addition of 20 μl of 1 M Tris-HCl solution and 15 min of centrifugation at 21,000×g. Upon the formation of precipitate, the supernatant was removed and placed in a new tube, and 100 μl of absolute isopropanol was added to the solution. The solution was centrifuged at 21,000×g for 15 min and the supernatant was discarded. Then, 200 μl of 70% ethanol was added and the solution centrifuged again at 21,000×g for 15 min. The supernatant was again discarded and the samples were oven-dried at 57 °C. To resuspend the DNA, 10 μl of Milli-Q water (Millipore, MA, USA) was added and the material was incubated at 37 °C for 30 min. The extracted DNA was quantified in a NanoDrop spectrophotometer (Thermo Fisher Scientific, CA, USA).
Preliminary PCR (step 2)
The NNC1F primer labeled with FAM fluorophore and the NC2R primer were also employed for the amplification of DNA of the ribosomal ITS-2 region (Table 1), using the amplification protocol described under item “Preliminary PRC (step 2).”
Analysis of fragments (step 3)
The methodology of DNA fragment analysis by capillary electrophoresis was chosen to develop and standardize the nematode identification test at the genus level.
In order to standardize the expected size of the fragments originating from each of the genera, DNA molecules were synthesized based on ITS-2 reference sequences retrieved from GenBank of the following parasite species prevalent in domestic ruminants herds in Brazil (Araújo and Lima 2005; Domingues et al. 2013; Ahid et al. 2008): H. contortus (AY647245), H. placei (X78812), C. oncophora (X83561), T. circumcincta (JF747153), O. ostertagi (JF747154), T. axei (EF427622), O. radiatum (AJ006149), and C. ovina (Y10789). The expected fragment sizes were entered into the GeneMapper software for use as reference.
The size of the fragments and the efficiency of the designed primers were tested (n = 190) using the synthetic sequences of the ITS-2 region of the seven genera of gastrointestinal parasitic nematodes of cattle, sheep, and goats most prevalent in Brazil.
The products amplified from the synthetic molecules or from biological samples in the preliminary PCR (step 2) were separated by capillary electrophoresis in an ABI Prism 3130 Genetic Analyzer (Thermo Fisher Scientific, CA, USA). The electrophoresis results were analyzed using GeneMapper software (Thermo Fisher Scientific, CA, USA) and the regions of the fluorescence peaks of the fragments of each genus were recorded and annotated.
Dilutions were made using Milli-Q water series from the synthetic sequences of each genus, and a fragment analysis was performed to examine the sensitivity of the test. Concentrations of 1 to 10−7 ng/μl of each synthetic sequence were tested in duplicate for each genus, making a total of 128 assays.
The sensitivity of the test was also confirmed in an assay in which the eggs were isolated individually in each well of a sequencing plate to extract the DNA from these specimens (n = 95). The extracted DNA was subjected to molecular identification of the genus by means of fragment analysis and then by sequencing the ITS-2 region in order to (i) verify the functioning of the test with DNA extracted from only one egg (evaluating the sensitivity of the test) and (ii) compare the proportion of genera identified by the fragment analysis and DNA sequencing methodologies. Thus, the proportion of each genus was calculated by dividing the peak height for each genus by the sum of the heights of all the fluorescence peaks observed in the fragment analysis performed with the GeneMapper software (Thermo Fisher Scientific, CA, USA).
To test the quantitative nature of this method, samples were prepared with mixtures containing known proportions of DNA of each of the studied genera, i.e., Cooperia spp., Haemonchus spp., Trichostrongylus spp., and Oesophagostomum spp. Different sets of mixtures were tested: with two, three, and the four genera and each blend with different proportions. Tests were also performed to determine if there was a proportional variation in the fluorescence peak of each of the genera (n = 95).
Specimens from a pool of L3 larvae that were identified morphometrically, as described under item “Larvae,” were also evaluated. In that case, the intensity of each fluorescence peak was considered to be indicative of the proportion of each species in the mixture. Fragment analyses of this same sample were also performed to compare the techniques based on the proportions found for each genus.
Minisequencing (step 4)
The minisequencing methodology using SNaPShot® kits (Thermo Fisher Scientific, CA, USA) was chosen for the development of the nematode identification test at the species level. Its use was based on the single nucleotide polymorphisms (SNPs) observed among the different analyzed nematode species.
Primer design
Reference sequences of the ITS-2 marker of the main gastrointestinal nematode species of domestic ruminants (H. contortus—AY647245, H. placei—X78812, H. longistipes—AJ577461, H. similis—L04152, C. punctata—X83560, C. oncophora—X83561, C. surnabada—AJ000032, C. curticei—AJ000033, C. ovina—Y10789, O. asperum—JX188457, O. radiatum—AJ006149, O dentatum—AJ889569, O. ostertagi—JF747154, T. circumcincta—JF747153, T axei—EF427622, T. colubriformis—X78063, and T. capricola—JF276022) were obtained from the GenBank database and aligned using MEGA 6.0 software (Tamura et al. 2013) to identify the polymorphic sites used as diagnostic positions and to design the primers. All the sequences available in the GenBank database for each of the species were also examined to confirm the absence of intraspecific and/or interspecific variations in the diagnostic sites. The designed primers are listed in Table 1.
Specific minisequencing primers were designed to differentiate between two or more possible species, according to their fluorescence (Table 2). Primers anneal in the region immediately adjacent to the diagnostic site and present different sizes. Non-homologous poly (dC) ends (with 9, 7, 14, and 11 dCTP, respectively) were added to the 5′ ends in the SNAP-4, SNAP-5, SNAP-6, and SNAP-7 primers to make fragments of different sizes, so that they could be specifically differentiated by multiplexed capillary electrophoresis.
Minisequencing reaction
Unlabeled NNC1F and NC2R primers (Table 1) aligned with conserved regions between the abovementioned parasite species were used for the PCR that preceded the minisequencing reaction. The conditions of this PCR were the same as those described for the preliminary PCR of step 2.
The products of the preliminary PCR were used as templates for the minisequencing reaction (n = 32). To this end, enzymatic purification was performed in order to remove the unincorporated primers and dNTPs using ExoSAP PCR cleanup reagent (USB Europe GmbH, Germany) according to the manufacturer’s instructions.
The minisequencing reactions were prepared using a concentration of 0.7 μM of the specific primers for the diagnosis of each species and/or genus, the ready reaction mix of the ABI PRISM SNaPshot® Multiplex Kit, (Thermo Fisher Scientific, CA, USA), and DNA extracted from a biological sample, which was previously sequenced for species identification. The sequencing reactions were performed according to the manufacturer’s instructions. In addition to template DNA sample testing, negative controls (without DNA added to the reaction) and a positive control (oligonucleotide components of the kit) were also prepared.
After the minisequencing reaction, the samples were prepared for capillary electrophoresis by adding 1 μl of the purified extension product to 24.6 μl of formamide (Thermo Fisher Scientific, CA, USA) and 0.4 μl of the GeneScan LIZ 120 size standard of molecular weight (Thermo Fisher Scientific, CA, USA). Each sample was then subjected to capillary electrophoresis in an ABI 3130 Genetic Analyzer (Thermo Fisher Scientific, CA, USA) and analyzed using POP7 polymer and a 47-cm capillary tube (Thermo Fisher Scientific, CA, USA). The ABI GeneScan E5 run module was used. The electropherograms thus generated were analyzed using GeneMapper software (Thermo Fisher Scientific, CA, USA).
Sanger sequencing of individual samples for method validation
The molecular marker ITS-2 of the nuclear rRNA of individual larva specimens was sequenced through capillary electrophoresis using Sanger method as a parallel standard approach to validate species identification results obtained using the semiquantitative method proposed in this study. DNA amplification was performed using the primers NNC1F and NC2R (Table 1, adapted from Stevenson et al. 1995). PCR amplifications were conducted in a final volume of 25 μl, including 10.5 μl of Milli-Q water, 6.25 μl of GoTaq Hot Start 5× PCR buffer (2.5 mM MgCl 2) (Promega), 0.25 μl of each primer (10 μM), 2.5 μl of each dNTP (1 mM), 0.25 μl GoTaq Hot Start polymerase (Promega) (5 U/μl), 2.5 μl of MgCl2 25 mM (Promega), and 2.5 μl of template DNA (50 ng/μl). The amplification protocol was as follows: 95 °C for 2 min, 35 cycles at 94 °C for 30 s, at 55 °C for 30 s and at 72 °C for 1 min, and a final extension at 72 °C for 10 min. The PCR conditions were adapted from those described by Brasil et al. (2012).
The sequences were determined bi-directionally on at least two distinct amplicons for each sample using the BigDye Terminator v.3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, CA, USA) and the primers described above in an ABI 3130 automated DNA sequencer (Thermo Fisher Scientific, CA, USA). The forward and reverse sequences were edited to remove ambiguous bases using SeqScape software (Thermo Fisher Scientific, CA, USA). The edited sequences were aligned using MEGA 6.0 software (Tamura et al. 2013).
The sequences generated here were compared with those deposited in the GenBank database (Benson et al. 2014), using the Basic Local Alignment Search Tool (BLAST) algorithm (Zhang et al. 2000) for the correct identification of the species of the specimens used.
Statistical analysis
Initially, the measures were dichotomized to evaluate the ability of the two methods to detect the presence of nematodes genera (n = 171). The samples in which the presence of each nematode genera was detected were recorded for each method and for each nematode. The proportion of concordant classifications was then calculated, as was Cohen’s kappa coefficient (Sim and Wright 2005). Moreover, in view of the detection by the molecular method, the detection probabilities were also calculated using the morphological method.
The measured values were then considered, using Pearson’s correlation coefficient to quantify the correlation between the measures found by the two methods. The concordance correlation coefficient based on the U-statistic was used to quantify the concordance of the measurements, as described by Carrasco et al. (2013). Lastly, the mean and standard error of the differences between the measures produced by the two methods (molecular and morphological) were calculated.
All the analyses were performed using the R program (Core Team). The kappa coefficient was calculated using the psych package and the concordance correlation coefficient was calculated using the cccrm package (Revelle 2015; Carrasco and Puig Martinez 2015). The results presented here are calculated with 95% confidence intervals.
Results
Identification of nematode genera
The different genera of gastrointestinal nematodes analyzed here were identified by the size of the fragment generated using primers that anneal in conserved regions of gene. The expected fragment sizes based on the sequences deposited into the GenBank database are as follows: Oesophagostomum spp.—306 bp; Haemonchus spp.—321 bp; Chabertia spp.—325 bp; Trichostrongylus spp.—327 bp; Ostertagia spp.—328 bp; Cooperia spp.—331 bp; and Teladorsagia spp.—336 bp. The actual fragment sizes observed for each genus in GeneMapper software (Thermo Fisher Scientific, CA, USA) during standardization and calibration tests using DNA extracted from a pool of L3 individualized (n = 190) were the following: Oesophagostomum spp.—306 to 314 bp; Haemonchus spp.—321 to 322 bp; Trichostrongylus spp.—328 to 330 bp; and Cooperia spp.—328 to 333 bp.
Using the synthetic sequences (n = 128) from the seven genera, i.e., Cooperia sp., Haemonchus sp., Trichostrongylus sp., Teladorsagia sp., Chabertia sp., Ostertagia sp., and Oesophagostomum sp., it was observed that the test works semiquantitatively up to a DNA concentration of 10−4 ng/μl, since the serial dilution of the DNA sequences (from 1 ng/μl to 1 ng/μl × 10−4) causes a gradual decrease in the fluorescence peaks of the fragments. Lower concentrations did not result in the gradual appearance of fluorescence peaks (data not shown).
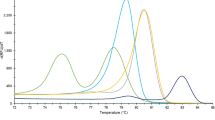
Analysis of samples of DNA extracted from individual Haemonchus spp., Cooperia spp., Trichostrongylus spp., and Oesophagostomum spp. eggs confirmed that a single egg can be used for the generic identification of gastrointestinal nematodes of ruminants (Fig. 1a–d). The amplification technique proved to be efficient for all the genera involved, as indicated by samples prepared from mixtures containing known proportions of egg DNA, such as the mixture of 70% of parasites of the genus Trichostrongylus spp. and 30% of the genus Haemonchus spp. In this case, it was observed that the proportion of peak heights of fluorescence for each species is proportional to the amounts of DNA of each of those genera in the samples. This finding suggests that the test should be semiquantitative.

Identification of genera based on fragment analysis of amplified DNA extracted from parasite eggs of: aOesophagostomum, bHaemonchus, cTrichostrongylus, and dCooperia. Gray bars indicate the bins referring to the fragment size patterns of the genera studied in the present work
The presence of the four different genera was also detected in DNA extracted from a pool of eggs from the field sample by means of fragment analysis by capillary electrophoresis. Table 3 shows the proportions of the species found by fragment analysis (n = 95). To validate this result, larvae from this same pool were individualized and subjected to Sanger sequencing of the ITS-2 region of each of them, thereby obtaining a quantitative reference of the proportion of genera and species in the sample. The proportion of these larvae found in an individual analysis was similar to the fluorescence quantification of each of the peaks of the fragment analysis.
The chi-square test revealed a significant difference (x2 = 1.120; p < 0.05; g.l. = 1) in the frequency resulting from each methodology between the species evaluated by the molecular (n = 95) and morphological (n = 100) methods. A comparison was also made of the results of the identification at the genus level of egg pools from field samples classified by morphological analysis and by the molecular method (fragment analysis) (n = 171). Thus, the intensity of each fluorescence peak in the fragment analysis was considered to be indicative of the proportion of each genus in the pool.
Qualitative detection of genera in L3
The concordance of detection of nematodes by the two techniques was high for all the genera (Oesophagostomum, Haemonchus, Trichostrongylus, and Cooperia), showing values of 0.84, 0.99, 0.84, and 0.90, respectively (Table 4). The kappa coefficient values for Oesophagostomum, Haemonchus, Cooperia, and Trichostrongylus were 0.56, 0.50, 0.66, and 0.18, respectively. The sensitivity and specificity of the techniques (considering the morphological technique as standard) for Oesophagostomum, Trichostrongylus, and Cooperia were, respectively, 072 and 087, 0.92 and 0.25, and 0.79 and 092 (Table 4).
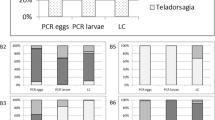
Figure 2a shows the percentage of negative samples (0%) in the detection of nematodes by the morphological or molecular techniques, or by the two methods simultaneously. Negative detections were obtained by the two techniques in 67, 0, 3, and 77% of the genera Oesophagostomum, Haemonchus, Trichostrongylus, and Cooperia, respectively, indicating a higher prevalence, in descending order, of the genera Haemonchus, Trichostrongylus, Oesophagostomum, and Cooperia. Nematodes of all the genera were detected in more analyses by the molecular than by the morphological method, i.e., fewer negatives were indicated by the former technique. The difference in negative detections between the techniques was 3, 1, 3, and 2% for the genera Oesophagostomum, Haemonchus, Trichostrongylus, and Cooperia, respectively (Fig. 2a). In 91% of the samples which showed divergent results in the detection of the genera, the quantitative percentage of the nematode was less than 10%, indicating its low content in the larval pool.

Percentage of negative samples (0%) for the detection of nematodes by morphological and molecular method, or by both techniques simultaneously (a). Comparison of the averages of identification of genera of gastrointestinal nematodes of sheep by morphological and molecular fragment analysis of 171 samples from L3 pools (b)
Quantitative detection of nematode genera in L3
The overall proportion of nematode genera in all the samples (n = 171) and the overall averages for morphological and molecular analysis of the genera Haemonchus Trichostrongylus, Oesophagostomum, and Cooperia are shown in Fig. 2b. The intra-sample average, which represents how much more a genus was detected in one sample than in another, differed by about 1% for all the nematodes. The correlation coefficients between samples presented in Table 5 for the genera Oesophagostomum, Haemonchus, Trichostrongylus, and Cooperia were, respectively, 0.77, 0.84, 0.85, and 0.80. The concordance between the data for the same sequence of genera was 0.75, 0.84, 0.88, and 0.68.
Nematode species identification
The preliminary PCR enabled the amplification of fragments of the ITS-2 gene of each species, and the minisequencing efficiency was tested in singleplex reactions (n = 32). Each reaction generated a peak of the expected color specific to its diagnostic site. The reactions were efficient in DNA extracted in all the nematode stages from eggs to adult worms. Samples of the species H. contortus, H. placei (Fig. 3), T. axei (Fig. 4), C. punctata, and O. radiatum (Fig. 5) were successfully identified.

Minisequencing of samples from different Haemonchus species. DNA was extracted from the isolates and subjected to preliminary PCR and fragment analysis, which indicated the presence of a sample of the genus Haemonchus (not shown). The original DNA concentration of all species evaluated was 50 ng/μl. Note the fluorescence peaks (y-axis) resulting from the minisequencing reaction of: a negative control (without DNA) (n = 2); b positive control of the kit (n = 2); c sample of DNA extracted from Haemonchus placei larvae in the presence of SNAP1 primer, which identifies this species by means of a peak of 21 bp of dCTP fluorescence (black) (n = 3); d sample of H. placei DNA in the presence of SNAP2 primer, which anneals only to H. contortus or H. longistipes, therefore leading, in this case, to the absence of a specific fluorescence peak (n = 3); e DNA sample extracted from a Haemonchus contortus larva in the presence of SNAP1 primer, which identifies this species by means of a peak of 21 bp of dCTP fluorescence (red) (n = 3) and f DNA sample extracted from a Haemonchus contortus larva in the presence of SNAP2 primer, which identifies this species by means of a peak of 25 bp of dCTP fluorescence (blue) (n = 3)

Minisequencing for identification of species of the genus Trichostrongylus. DNA was extracted from the isolates and subjected to preliminary PCR and fragment analysis, which indicated the presence of a sample of the genus Trichostrongylus (not shown). The original DNA concentration of all species evaluated was 50 ng/μl. The fluorescence peaks in this figure resulted from the minisequencing reaction of: a negative control (n = 2); b positive control of the kit (n = 2), and c DNA sample extracted from a Trichostrongylus colubriformis larva in the presence of SNAP7 primer, which identifies this species by means of a peak of 32 bp of dCTP fluorescence (black) (n = 3)

Minisequencing for identification of the genus Cooperia and the species Oesophagostomum radiatum. DNA was extracted from the isolates and subjected to preliminary PCR and fragment analysis, which indicated the presence of samples of the genera Cooperia and Oesophagostomum (not shown). The fluorescence peaks in this figure resulted from the minisequencing reaction of: a negative control (n = 2); b positive control of the kit (n = 1); c DNA sample extracted from a Cooperia punctata larva in the presence of SNAP8 primer, which identifies this species by means of peak of 21 bp of dCTP fluorescence (black) (n = 3), and d DNA sample extracted from an Oesophagostomum radiatum larva in the presence of SNAP10 primer, which identifies this species by means of a peak of 19 bp of dCTP fluorescence (blue) (n = 3)
Discussion
The PCR-based methods have increased in popularity in the scientific community (Amarante et al. 2017; Rashid et al. 2018). In addition, more and more, the researches seek to identification and quantification of the most abundant genera of strongylids in sheep and with a major focus on Haemonchus, which is regarded as the most pathogenic nematode in small ruminants (Elmahalawy et al. 2018). However, the simultaneous analysis of multiple nematode species is often hampered by the fact that techniques based on conventional or real-time PCR lose reproducibility and accuracy as the number of primers in the reaction mix (multiplex) increases, particularly due to the multiple possible interactions between them during the amplification of target DNA (Zarlenga et al. 2001; Höglund et al. 2013; Roeber et al. 2017a). The technique described in here consists of a simple and effective test for the diagnosis of nematodes parasites of ruminants individually or in mixed populations, through the amplification of ITS-2 region using a single universal primer pair followed by fragment analysis and minisequencing. Recently reported approaches based on deep nemabiome sequencing approach also explores this highly polymorphic DNA region of DNA to identify parasites but uses the deep sequencing strategy (Avramenko et al. 2015, 2017). Such methods use the Illumina MiSeq platform, similarly to metabarcoding strategies performed in microbial population studies.
Fragment analysis proved to be a cost- and time-effective technique to identify the genera Haemonchus, Cooperia, Oesophagostomum, and Trichostrongylus in the various assays carried out to validate the technique compared to other approaches. These assays demonstrated that the fragment sizes of the ITS-2 gene of these helminths are sufficiently polymorphic to differentiate these genera. Fragment size variations for the same genus may have occurred due to differences in alleles in the population and/or to the different configurations of the automated sequencers (three different sequencers were used for the technique to enable the observation of possible equipment-dependent variations). To correct such variations, the bins for GeneMapper software (Thermo Fisher Scientific, CA, USA) were adjusted according to the mean values and deviations found in all the assays.
The semiquantitative detection of each of the genera contained in the samples was also successfully performed, as shown in Table 3. Comparative tests of morphological and molecular analyses to determine the proportion of larvae using pool fragment analysis revealed a strong association between the values found by the two methods (Table 4). In this test, by adding up the height of the peaks found for each genus and assuming this value to be 100%, the approximate proportion of each genus in the pool was calculated based on the height of the peak corresponding to each of the genera. In some cases, the presence of genera undetected in the morphological analysis was detected in the molecular analysis, demonstrating the greater sensitivity of the molecular technique. This characteristic is fundamental when conducting research into the efficacy of an active substance against the main species of nematodes, and particularly when investigating the efficacy of anthelmintic during monitoring of a proposed control program in production systems.
Studies based on comparisons of morphological and molecular identification (Roeber et al. 2017a, b; Ljungström et al. 2018) of infectious parasites use the multiplex PCR method. The advantage of the technique proposed here stems from the fact that it requires only the use of a pair of primers for the genus-specific identification of the most prevalent gastrointestinal nematodes in tropical herds. Thus, we propose a fast and cost-effective method based on the molecular identification of these parasites.
A significant advantage of the fragment analysis technique proposed here for the genera identification is that is can be carried out using a pool of specimens in only one assay. In contrast, techniques that are frequently used to identify parasites, such as morphological identification and DNA sequencing, require individual evaluation of specimens. In addition, we found that the primers did not exhibit greater effectiveness with one genus than with the others, which is a prerequisite for studying the proportion of each genus in the sample.
The sensitivity and specificity of detection by the methods were high for all the genera except Haemonchus, for which no value was recorded because the parasite was present in 99% of the cases. Specificity for Trichostrongylus was low because only a few negatives were found in both samples, and this also reduced the kappa index. According to the classification proposed by Landis and Koch (1977), the kappa index showed intermediate values for Oesophagostomum, Haemonchus, and Cooperia and low for Trichostrongylus (0.18). The reason for this was that most of the samples, especially in the case of Trichostrongylus and Haemonchus, tested positive by both methods, with no marked predominance of negative-negative cases. Thus, the coefficient ended up “ignoring” the possibility of chance coincidence (strongly related to positive-positive cases), so the level of concordance was lower.
From the epidemiological and clinical standpoints, it is not enough merely to detect parasitic genera or species, given the differences between their pathogenicities. The correlation indices between the percentages of genera were strong (Mukaka 2012). Concordance between percentages was also high, especially when the percentage of the genus was high in the larval pool, as in the case of Haemonchus and Trichostrongylus. Therefore, the percentage of larvae in the pool affected not only the qualitative but also the quantitative diagnosis. This fact decreased the concordance of the values for Oesophagostomum and Cooperia, which showed median values below 3% of the total percentage. Nevertheless, the general average of the percentages and the intra-sample average were very close, confirming that the greater the number of evaluations, the smaller the chance of error in the case of morphological evaluations. This is especially important when the percentage of nematodes is low. Using the morphological technique, we evaluated 100 L3 per sample. This was an extremely expensive undertaking, considering the large amount of material to be diagnosed in an ordinary laboratory of parasitology.
The molecular technique proved to be sensitive for the detection of amplified DNA from pools of eggs, larvae, or adult worms. This sensitivity for detection in small initial amounts of DNA is an essential differential to render the technique faster and more efficient. The technique is fast because the identification of genera based on DNA extracted from eggs does not require the coproculture step to obtain larvae, and it is efficient because eggs contain less DNA than samples with larger numbers of cells, such as larvae and adult worms.
Another differential of this molecular technique is the fact that it does not need a set of primers for the identification of genera as used in the reaction mix (multiplex). A set of primers can lead to different levels of amplification efficiency, making semiquantitative analysis difficult. Moreover, using just one pair of primers as here increases the reproducibility and accuracy of the method.
The minisequencing technique was proposed for the identification of parasites at species level complementing the fragment analysis. The designed primers adjacent to the genus- or species-specific SNPs allowed for the molecular identification of parasites whose DNA was extracted individually. As with fragment analysis, singleplex minisequencing reactions were effective in samples extracted at different stages of development. Moreover, in principle, the technique can also be applied to pools of parasites, ensuring its applicability to species diagnosis. Thus, minisequencing was effective in identifying the species H. contortus and H. placei, C. punctata and C. curticei, T. colubriformis, and O. radiatum. It was also effective in differentiating specimens of the genus Haemonchus and Trichostrongylus. Different from metabarcoding (Avramenko et al. 2017), this is an interesting alternative when the full amplicon sequencing is not required for the objectives of the study. The accurate identification of different species, as well as knowledge about the epidemiology of parasitic gastroenteritis, is essential for the establishment of sustainable parasite control strategies.
The ability to simultaneously investigate different regions of DNA and the high potential for automation and scalability make this technique an extremely advantageous alternative to morphological diagnosis, especially when several species need to be identified. This is the first description of the use of the minisequencing technique for the diagnosis of parasitic helminth species of domestic ruminants. This technique has been used for the identification of species of microorganisms (Dalmasso et al. 2010; Bouakaze et al. 2010; Huang et al. 2011, 2012). Furthermore, Reale et al. (2008) demonstrated that the amount of DNA from a species present in a sample can be estimated based on the size of peaks generated in the minisequencing multiplex reaction. This author was able to establish a correlation between the height of a peak and the sum of the height of the peaks and to estimate the proportion of a given species in the analyzed sample as shown here.
The concomitant application of fragment analysis and minisequencing techniques thus enabled the identification of species that cause significant economic losses to Brazil’s livestock industry. The proposed methodology, which consists of two analytical steps, is fast, high scalable and affordable, and can contribute to the choice of suitable drugs for use in helminth control programs. Thus, it can contribute to reduce financial losses incurred by breeders as a result of the high costs of anthelmintic drugs, and also produce scientific knowledge helpful for monitoring and preventing these parasitic diseases in livestock.
Conclusions
The technique of fragment analysis of the ITS-2 gene proved to be useful for the semiquantitative identification of the genera Oesophagostomum, Haemonchus, Trichostrongylus, and Cooperia from samples containing low DNA concentrations. Moreover, the technique can also be employed to identify species of the genera Ostertagia and Teladorsagia, using the same pair of primers described here. The SNaPshot® minisequencing method enabled the identification of the species H. contortus, H. placei, C. punctata/C. curticei, T. colubriformis, and O. radiatum. Twelve species of ruminant parasitic nematodes (H. contortus, H. placei, H. longistipes, C. curticei, C. punctata, C. oncophora/C. surnabada, C. ovina, O. asperum, O. radiatum, O. ostertagi, T. axei, and T. colubriformis) can be identified with this technique, using the primers described here.
The proposed method offers advantages over coproculture technique for the identification of parasitic nematodes of the gastrointestinal tract of ruminants, such as high specificity, precision, reproducibility, potential for automation, and easy interpretation of results for the identification of the main nematode parasites. The method is not only more specific, semiquantitative, and faster—only 2 days for diagnosis—but also less time-consuming and more affordable, especially for low EPG counts.
References
Ahid SMM, Suassuna ACD, Maia MB, De Mendonça Costa VM, Soares HS (2008) Parasitos gastrintestinais em caprinos e ovinos da região oeste do Rio Grande do Norte, Brasil. Ciênc Anim Bras 9:212–218
Amarante MRV, Santos MC, Bassetto CC, Amarante AFT (2017) PCR primers for straightforward differentiation of Haemonchus contortus, Haemonchus placei and their hybrids. J Helminthol 91:757–761
Araújo RN, Lima WS (2005) Infecções helmínticas em um rebanho leiteiro na região Campo das Vertentes de Minas Gerais. Arq Bras Med Vet Zootec 57:186–193
Avramenko RW, Redman EM, Lewis R, Yazwinski TA, Wasmuth JD, Gilleard JS (2015) Exploring the gastrointestinal “nemabiome”: deep amplicon sequencing to quantify the species composition of parasitic nematode communities. PLoS One 10:1–18
Avramenko RW, Redman EM, Lewis R, Bichuette MA, Palmeira BM, Yazwinski TA, Gilleard JS (2017) The use of nemabiome metabarcoding to explore gastro-intestinal nematode species diversity and anthelmintic treatment effectiveness in beef calves. Int J Parasitol 47:893–902
Avramenko RW, Bras A, Redman EM, Woodbury MR, Wagner B, Shury T, Liccioli S, Windeyer MC, Gilleard JS (2018) Exploring the Gastrointestinal “Nemabiome”: Deep Amplicon Sequencing to Quantify the Species Composition of Parasitic Nematode Communities. Parasit Vectors 11:299 – 311
Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2014) GenBank. Nucleic Acids Res 42:D32–D37
Blouin MS (2002) Molecular prospecting for cryptic species of nematodes: mitochondrial DNA versus internal transcribed spacer. Int J Parasitol 32:527–531
Bott NJ, Campbell BE, Beveridge I, Chilton NB, Rees D, Hunt PW, Gasser RB (2009) A combined microscopic-molecular method for the diagnosis of strongylid infections in sheep. Int J Parasitol 39:1277–1287
Bouakaze C, Keyser C, De Martino SJ, Sougakoff W, Veziris N, Dabernat H, Ludes B (2010) Identification and genotyping of Mycobacterium tuberculosis complex species by use of a SNaPshot minisequencing-based assay. J Clin Microbiol 48:1758–1766
Brasil BS, Nunes RL, Bastianetto E, Drummond MG, Carvalho DC, Leite RC, Molento MB, Oliveira DA (2012) Genetic diversity patterns of Haemonchus placei and Haemonchus contortus populations isolated from domestic ruminants in Brazil. Int J Parasitol 425:469–479
Carrasco JL, Puig Martinez J (2015) cccrm: concordance correlation coefficient for repeated (and non-repeated) measures. R package version 1.2.1. https://cran.r-project.org/web/packages/cccrm/index.html
Carrasco JL, Phillips BR, Puig-Martinez J, King TS, Chinchilli VM (2013) Estimation of the concordance correlation coefficient for repeated measures using SAS and R. Comput Methods Prog Biomed 109:293–304
Charlier J, Thamsborg SM, Bartley DJ, Skuce PJ, Kenyon F, Geurden T, Hoste H, Williams AR, Sotiraki S, Höglund J, Chartier C, Geldhof P, Van Dijk J, Rinaldi L, Morgan ER, Von Samson-Himmelstjerna G, Vercruysse J, Claerebout E (2018) Mind the gaps in research on the control of gastrointestinal nematodes of farmed ruminants and pigs. Transbound Emerg Dis 65:217–234
Coelho WAC, Ahid SMM, Vieira LS, Fonseca ZAAS, Silva IP (2010) Resistência anti-helmíntica em caprinos no município de Mossoró, RN. Ciênc Anim Bras 11:589–599
Cort WW, Ackert JE, Augustine DL, Payne FK (1922) Investigations on the control of hooworm disease. II. The description of an apparatus for isolating infective hookworm larvae from soil. Am J Hyg 2:1–16
Dalmasso A, Rantsiou K, Cocolin L, Bottero MT (2010) Development of a biomolecular assay for the identification of Listeria at species level. Foodborne Pathog Dis 7:565–571
Dobson RJ, Le Jambre L, Gill JH (1996) Management of anthelmintic resistance: inheritance of resistance and selection with persistent drugs. Int J Parasitol 26:993–1000
Domingues LF, Giglioti R, Feitosa KA, Fantatto RR, Rabelo MD, De Sena Oliveira MC, Bechara GH, Oliveira GP, Braioni Junior W, Souza Chagas AC (2013) In vitro and in vivo evaluation of the activity of pineapple (Ananas comosus) on Haemonchus contortus in Santa Inês sheep. Vet Parasitol 197:263–270
Elmahalawy ST, Halvarsson P, Skarin M, Höglund J (2018) Droplet digital polymerase chain reaction (ddPCR) as a novel method for absolute quantifica of major gastrointestinal nematodes in sheep. Vet Parasitol 261:1–8.
Gasser RB (2006) Molecular tools—advances, opportunities and prospects. Vet Parasitol 136(2):69–89
Gasser RB, Monti JR, Zhu X, Chilton NB, Hung GC, Guldberg P (1997) Polymerase chain reaction-linked single-strand conformation polymorphism of ribosomal DNA to fingerprint parasites. Electrophoresis 18:1564–1566
Gilleard JS (2006) Understanding anthelmintic resistance: the need for genomics and genetics. Int J Parasitol 36:1227–1239
Gordon NM, Withlock HV (1939) A new technique for counting nematode eggs in sheep faeces. J Counc Sci Ind Res 12:50–52
Hansen J, Perry B (1994) The epidemiology, diagnosis and control of helminth parasites of ruminants. International Laboratory on Animal Diseases, Nairobi, pp 83–90
Höglund J, Engström A, Von Samson-Himmelstjerna G, Demeler J, Tydén E (2013) Real-time PCR detection for quantification of infection levels with Ostertagia ostertagi and Cooperia oncophora in cattle faeces. Vet Parasitol 197:251–257
Huang CH, Chang MT, Huang MC, Lee FL (2011) Application of the SNaPshot minisequencing assay to species identification in the Lactobacillus casei group. Mol Cell Probes 25:153–157
Huang CH, Chang MT, Huang MC, Wang LT, Huang L, Lee FL (2012) Discrimination of the Lactobacillus acidophilus group using sequencing, species-specific PCR and SNaPshot mini-sequencing technology based on the recA gene. J Sci Food Agric 92:2703–2708
Kaplan RM (2004) Drug resistance in nematodes of veterinary importance: a status report. Trends Parasitol 20:477–481
Landis JR, Koch GG (1977) The measurement of observer agreement for categorical data. Biometrics 33:159–174
Learmount J, Conyers C, Hird H, Morgan C, Craig BH, Von Samson-Himmelsterna G, Taylor M (2009) Development and validation of real-time PCR methods for diagnosis of Teladorsagia circumcincta and Haemonchus contortus in sheep. Vet Parasitol 166:268–274
Ljungström S, Melville L, Skuce PJ, Höglund J (2018) Comparison of four diagnostic methods for detection and relative quantification of Haemonchus contortus eggs in feces samples. Front Vet Sci 4:1–7
Mukaka MM (2012) A guide to appropriate use of correlation coefficient in medical research. Malawi Med J 24:69–71
Nielsen MK, Peterson DS, Monrad J, Thamsborg SM, Olsen SN, Kaplan RM (2008) Detection and semi-quantification of Strongylus vulgaris DNA in equine faeces by real-time quantitative PCR. Int J Parasitol 38:443–453
Rashid MH, Gebrekidan H, Jabbar A (2018) Multiplexed-tandem PCR (MT-PCR) assay to detect and differentiate gastrointestinal nematodes of alpacas. Parasit Vectors 11:370
RAUE K, Heuer L, BÖHM C, WOLKEN S, EPE C, STRUBE C (2017) 10-year parasitological examination results (2003 to 2012) of faecal samples from horses, ruminants, pigs, dogs, cats, rabbits and hedgehogs. Parasitol Res 116:3315–3330
Reale S, Campanella A, Merigioli A, Pilla F (2008) A novel method for species identification in milk and milk-based products. J Dairy Res 75:107–112
Redman EM, Queiroz C, Bartley DJ, Levy M, Avramenko RW, Gilleard JS (2019) Validation of ITS-2 rDNA nemabiome sequencing for ovine gastrointestinal nematodes and its application to a large scale survey of UK sheep farms. Vet Parasitol 275
Revelle W (2015) Psych: procedures for personality and psychological research. Northwestern University, Evanston http://CRAN.R-project.org/package=psychVersion=1.5.8
Roberts FHS, O'Sullivan PJ (1950) Methods for egg counts and larval cultures for strongyles infesting the gastro-intestinal tract of cattle. Crop Pasture Sci 1:99–102
Roeber F, Jex AR, Campbell AJ, Campbell BE, Anderson GA, Gasser RB (2011) Evaluation and application of a molecular method to assess the composition of strongylid nematode populations in sheep with naturally acquired infections. Infect Genet Evol 11:849–854
Roeber F, Jex AR, Campbell AJ, Nielsen R, Anderson GA, Stanley KK, Gasser RB (2012) Establishment of a robotic, high-throughput platform for the specific diagnosis of gastrointestinal nematode infections in sheep. Int J Parasitol 42:1151–1158
Roeber F, Hassan EB, Skuce P, Morrison A, Claerebout E, Casaert S, Homer DR, Firestone S, Stevenson M, Smith L, Larsen J (2017a) An automated, multiplex-tandem PCR platform for the diagnosis of gastrointestinal nematode infections in cattle: an Australian-European validation study. Vet Parasitol 30:62–75
Roeber F, Morrison A, Casaert S, Smith L, Claerebout E, Skuce P (2017b) Multiplexed-tandem PCR for the specific diagnosis of gastrointestinal nematode infections in sheep: an European validation study. Parasit Vectors 10:226–237
Salgado JA, Santos CP (2016) Overview of anthelmintic resistance of gastrointestinal nematodes of small ruminants in Brazil. Rev Bras Parasitol Vet 25:3–17
Silvestre A, Humbert JF (2000) A molecular tool for species identification and benzimidazole resistance diagnosis in larval communities of small ruminant parasites. Exp Parasitol 95:271–276
Sim J, Wright CC (2005) The kappa statistic in reliability studies: use, interpretation, and sample size requirements. Phys Ther 85:257–268
Stevenson LA, Chilton NB, Gasser RB (1995) Differentiation of Haemonchus placei from H.contortus (Nematoda: Trichostrongylidae) by the ribosomal DNA second internaltranscribed spacer. Int J Parasitol 25:483–488
Sutherland IA, Leathwick DM (2011) Anthelmintic resistance in nematode parasites of cattle: a global issue? Trends Parasitol 27:176–181
Tamura K, Stecher G, Peterson D, Filipski IA, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Torres-Acosta JFJ, Mendoza-de-Gives P, Aguilar-Caballero AJ, Cuéllar-Ordaz JA (2012) Anthelmintic resistance in sheep farms: update of the situation in the American continent. Vet Parasitol 189:89–96
Ueno H, Gonçalves PC (1998) Manual para diagnóstico da helmintoses de ruminantes, 4th edn. Japan International Cooperation Agency, p 72
Van Wyk JA, Mayhew E (2013) Morphological identification of parasitic nematode infective larvae of small ruminants and cattle: a practical lab guide. Onderstepoort J Vet Res 80:539–553
Van Wyk JA, Cabaret J, Michael L (2004) Morphological identification of nematode larvae of small ruminants and cattle simplified. Vet Parasitol 119:277–306
Várady JA, Cudekova P, Corba J (2007) In vitro detection of benzimidazole resistance in Haemonchus contortus: egg hatch test versus larval development test. Vet Parasitol 149:104–110
Von Samson-Himmelstjerna G (2006) Molecular diagnosis of anthelmintic resistance. Vet Parasitol 136:99–107
Zarlenga DS, Barry Chute M, Gasbarre LC, Boyd PC (2001) A multiplex PCR assay for differentiating economically important gastrointestinal nematodes of cattle. Vet Parasitol 97:201–211
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7:203–214
Acknowledgments
The authors would like to thank Ricardo Land Lomardo, the other farm owners, and Letícia Vidal Cruz for their technical support.
Funding
Financial support for this study was provided by the Brazilian research funding agencies CNPq (National Council for Scientific and Technological Development) (INCT 573899/2008-8), FAPEMIG (Minas Gerais State Research Foundation) (INCT APQ-0084/08), FAPERJ (Rio de Janeiro Research Foundation), and CAPES (Coordination for the Improvement of Higher Education Personnel) (Finance Code 001).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Section Editor: Georg von Samson-Himmelstjerna
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Santos, L.L., Salgado, J.A., Drummond, M.G. et al. Molecular method for the semiquantitative identification of gastrointestinal nematodes in domestic ruminants. Parasitol Res 119, 529–543 (2020). https://doi.org/10.1007/s00436-019-06569-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-019-06569-3