
Abstract
Chagas disease is a tropical parasitic disease caused by the protozoan Trypanosoma cruzi, which affects about ten million people in its endemic regions of Latin America. After the initial acute stage of infection, 60–80% of infected individuals remain asymptomatic for several years to a lifetime; however, the rest develop the debilitating symptomatic stage, which affects the nervous system, digestive system, and heart. The challenges of Chagas disease have become global due to immigration. Despite well-documented dietary changes accompanying immigration, as well as a transition to a western style diet in the Chagas endemic regions, the role of host metabolism in the pathogenesis of Chagas disease remains underexplored. We have previously used a mouse model to show that host diet is a key factor regulating cardiomyopathy in Chagas disease. In this study, we investigated the effect of a high-fat diet on liver morphology and physiology, lipid metabolism, immune signaling, energy homeostasis, and stress responses in the murine model of acute T. cruzi infection. Our results indicate that in T. cruzi-infected mice, diet differentially regulates several liver processes, including autophagy, a stress response mechanism, with corresponding implications for human Chagas disease patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chagas disease is a neglected tropical disease caused by the protozoan parasite Trypanosoma cruzi (Tanowitz et al. 1992; Grayson 2010). Symptoms of T. cruzi infection vary but include heart disease, hepatomegaly, and mega syndromes of the gastrointestinal tract. An estimated ten million people infected with T. cruzi reside in Central and South America (Bonney 2014). Globalization due to immigration has increased the cases of Chagas disease in developed countries including the USA. In North America alone, more than 370,000 individuals are currently living with T. cruzi infection (Tanowitz et al. 2011). The dietary changes accompanying immigration, plus the social and economic changes occurring over the last six decades in Chagas endemic countries, including a transition to a western style diet, have had a significant metabolic impact in the Chagas disease population. However, how host metabolism affects Chagas disease progression and pathogenesis is only beginning to be understood.
Our recent studies have primarily focused on understanding the impact of diet and metabolic changes on the pathogenesis of chagasic cardiomyopathy caused by T. cruzi infection. We have shown that T. cruzi has a high affinity for lipids and lipoproteins, that its rate of invasion depends on host cholesterol levels, and that it alters host lipid metabolism to suit its nutritional requirements, replication, and survival (Johndrow et al. 2014; Combs et al. 2005). We also recently demonstrated that in a murine model of Chagas disease, T. cruzi-infected mice that were fed on a high-fat diet (HFD) had increased survival compared to mice fed on a regular diet (RD) (Nagajyothi et al. 2014; Brima et al. 2015). Adipose tissues of the infected mice play a major role in reducing parasite load and inflammation in HFD fed mice, which may contribute to the increased survival rate during acute T. cruzi infection. In mice fed a RD, we found that T. cruzi infection causes significant lipolysis (60%), which may contribute to cardiac lipidopathy and high parasite loads during acute infection (Nagajyothi et al. 2014). In contrast, HFD ameliorated the effects of infection of adipose tissue and reduced adipocyte lipolysis (Nagajyothi et al. 2014). Infected mice fed on a HFD also showed improved cardiac pathology compared to infected mice fed on a RD [7]. Together, all these findings suggest that Chagas disease regulates host energy metabolism and that lipid content in the diet may play a pivotal role in the pathogenesis of this disease.
The liver is a key metabolic organ, which governs body energy metabolism by acting as a hub to metabolically connect various tissues, including cardiac muscle and adipose tissue. Therefore, the function of the liver is greatly regulated by diet and circulating lipoprotein levels, and vice versa. Furthermore, it has been reported that acute Chagas disease causes liver enlargement (Bern et al. 2011; Shikanai-Yasuda and Carvalho 2012) and that Chagas disease can be transmitted through liver transplantation (Rodriguez-Guardado et al. 2015), indicating that the liver harbors the pathogen. Thus, it is likely that the liver plays an important role in the pathogenesis of Chagas disease and may regulate the survival of mice during acute T. cruzi infection in a diet-dependent manner. However, thus far, the role of the liver in Chagas disease has been investigated only in regard to its function in the clearance of blood trypomastigotes (Sardinha et al. 2010). In this study, we focused on understanding the role of the liver in lipid metabolism, inflammation, oxidative stress, and energy homeostasis during acute T. cruzi infection in a murine model of Chagas disease fed on different diets.
It has been demonstrated that the liver is highly dependent on autophagy for normal function and to prevent the development of disease states (Schneider and Cuervo 2014). Autophagy is a key part of the cellular stress response allowing the cell to cope with nutrient starvation and anoxia, both of which are conditions commonly associated with infection (Russell et al. 2014). It has been shown that the fat content of the diet regulates autophagic flux in different organs (Chu et al. 2015; Hsu et al. 2015; Liu et al. 2015). Thus, in this study, we evaluated the role of autophagy in the liver during acute T. cruzi infection in the context of different diets in liver.
Methods
Experimental animal model
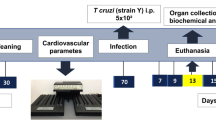
T. cruzi (Brazil strain) was maintained by passage in C3H/Hej mice (Jackson Laboratories, Bar Harbor, ME). CD-1 mice (8–10-week-old male mice purchased from Jackson Laboratories) were intraperitoneally (i.p.) infected with 5 × 104 trypomastigotes (Combs et al. 2005). Mice were maintained on a 12-h light/dark cycle. Mice were randomly divided into two groups (n = 30 per group) and fed on either HFD (60% fat) or RD (10% fat) (Research Diets, Inc., New Brunswick, NJ), starting at the day of infection. Mice (uninfected) fed on either HFD (n = 30) or RD (n = 30) were used as respective diet controls in all the experiments (Nagajyothi et al. 2014). The experiment was duplicated using the same numbers of mice in all groups.
Quantitative determination of parasite load in liver samples
Livers were collected from mice on days 5, 10, 20, and 30 post-infection (pi) and stored at −80 °C. The parasite load was quantified by a quantitative real-time polymerase chain reaction (qPCR) analysis employing PCR SYBR Green Master Mix (RT2 SYBR® Green qPCR Mastermix, Qiagen, CA) and an iQ5 LightCycler (Bio-Rad). The protocol for the isolation of DNA, preparation of standard curves for host and epimastigote DNA, and qPCR analysis was followed as previously published (Nagajyothi et al. 2014).
Analysis of liver cholesterol
Cholesterol levels were quantified in the livers of mice at day 30 pi using a colorimetric assay kit, and samples were prepared and assayed following the manufacturer’s protocol (Total cholesterol colorimetric assay kit, Cell Biolabs Inc., CA).
Immunoblot analysis
Liver lysates were prepared as previously described (Nagajyothi et al. 2014). An aliquot of each sample (40 μg protein) was subjected to a 4–15% gradient SDS-PAGE (except 14% gel used for LC3), and the proteins were transferred to nitrocellulose filters for immunoblot analysis. p-AMPK beta-specific rabbit monoclonal antibody (1:1000 dilution, Ser108, Cell Signaling), AMPK beta-specific rabbit monoclonal antibody (1:1000 dilution, Cell Signaling), ABCA1-specific mouse monoclonal antibody (1:500 dilution, AB.H10, Abcam), p-mTOR-specific rabbit (1:1000 dilution, Ser2448, Cell Signaling), LC3 B1/2-specific mouse (1:1000 dilution, M 186–3, MBL), p-62-specific rabbit (1:1000 dilution, PM045, MBL), fatty acid synthase-specific rabbit monoclonal antibody (1:1000 dilution, C20G5, Cell Signaling), SOD1-specific mouse monoclonal antibody (1:1000 dilution, 71G8, Cell Signaling), UCP3-specific rabbit monoclonal antibody (1:1000 dilution, D6J8K, Cell Signaling), TNF-alpha-specific rabbit polyclonal antibody (1:2000 dilution, AB6671, Abcam), and interferon gamma-specific rabbit monoclonal antibody (1:1000 dilution, EPR1108, Abcam) were used as primary antisera. Horseradish peroxidase-conjugated goat anti-mouse immunoglobulin (1:2000 dilution, Thermo Scientific) or horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin (1:2000 dilution, Thermo Scientific) were used to detect specific protein bands (explained in figure legends) using a chemiluminescence system (Nagajyothi et al. 2014). GDI (1: 10,000 dilution, 71-0300, and rabbit polyclonal, Invitrogen, CA) and a secondary antibody horseradish peroxidase conjugated goat anti-rabbit (1:2000 dilution, Amersham Biosciences) was used to normalize protein loading.
Polymerase chain reaction array
An RT2 Profiler (SA Biosciences, Valencia, CA) custom designed PCR array for mouse genes involved in LDL internalization, cholesterol metabolism, fatty acid and triglyceride metabolism, glucose metabolism, and inflammatory signaling was used to analyze gene expression. Data analysis was performed normalized to the expression of 18S RNA using the ΔΔCT method according to the manufacturer’s protocol (Qiagen, CA) (Nagajyothi et al. 2014).
Immunohistochemistry and IFA
Freshly isolated tissues were fixed with phosphate-buffered formalin overnight and then embedded in paraffin wax. Hematoxylin and eosin (H&E) staining was performed, and the images were captured as previously published (Nagajyothi et al. 2014). Filipin staining was done to analyze free cholesterol levels in the livers as previously demonstrated (Johndrow et al. 2014). Immunohistochemical analysis was performed on the formalin-fixed liver sections to analyze the presence of infiltrated macrophages and Kupffer cells using F4/80-specific rabbit monoclonal antibody (1:250 dilution, AB111101, Abcam) as demonstrated earlier (Combs et al. 2005).
Measurement of live triglyceride levels by 1H-MRS analysis
Magnetic resonance imaging (MRI) of mice was performed at day 30 pi as previously mentioned (Nagajyothi et al. 2014). Immediately after MR imaging, localized 1H-MRS was performed using a respiratory-gated PRESS sequence (TE = 15 ms, TR ∼ 10 s, spectral bandwidth = 4006 Hz) with a 90° Shinnar-Le Roux pulse for excitation and Mao-type refocusing pulses. An ∼20-ml voxel was positioned in liver. Volume selective shimming of the voxel was done manually using the same PRESS sequence resulting in water signal line width (FWHM) less than 40 Hz. Spectrum was acquired with 16 averages without water suppression. All spectra were analyzed in the frequency domain using Agilent VnmrJ integration routine. Hepatic lipid contents were expressed as ratios of the integral of the triglyceride methylene peak at relative to that of the water peak.
Statistical analysis
Immunoblot, immunofluorescence, and quantification of parasite load studies were performed at least three times, and representative data are presented in the figures. Data were pooled and statistical analysis was performed using a Student’s t-test (Microsoft Excel) as appropriate and significance differences were determined as p values between <0.05 and <0.005 as appropriate. Gene array analyses were done in duplicate as described earlier (Nagajyothi et al. 2014).
Results
We analyzed the role of the liver in the pathogenesis of acute Chagas disease by various methods, including immunoblotting and qPCR analysis, to further decipher the mechanisms of the protective effect of HFD on the pathogenesis of acute Chagas disease.
HFD increases hepatic parasite load during acute T. cruzi infection
We used qPCR to analyze the effect of HFD on the parasite load in the liver at different time points (days 5, 10, 20, and 30 pi) of acute infection (Nagajyothi et al. 2014). We found that HFD fed mice had increased hepatic parasite levels, which were 2-fold (on days 5 and 10 pi) and 5-fold (on day 30 pi) greater in HFD fed mice compared to RD fed mice (Fig. 1).

High-fat diet (HFD) increases hepatic parasite loads in mice during acute T. cruzi infection. qPCR analysis demonstrated a significant increase of parasites in the livers of HFD fed infected mice compared to RD fed infected mice at different time points of acute infection (n = 5). The parasite loads were significantly higher (17-, 2-, and 5-fold, respectively) at days 5, 10, and 30 post-infection (pi). p ≤ 0.005, represented by black stars. Bars represent mean values of the data with standard error of the mean (SEM) as vertical lines. RD regular diet, HFD high-fat diet
Cholesterol accumulates in hepatocytes during acute infection
Filipin staining followed by immunofluorescence analysis (IFA) demonstrated significant cholesterol accumulation in various organs and tissues, including the livers of infected mice (supplemental Fig. 1) (Johndrow 2014). Consistent with the IFA, colorimetric measurements also showed a significant increase in hepatic cholesterol levels during acute infection (Fig. 2a). Both HFD and RD fed infected mice showed approximately 50% increase in hepatic cholesterol levels compared to RD fed uninfected mice. Infected HFD fed mice showed slightly increased (average) cholesterol levels in the liver compared to infected RD fed mice; however, there was no statistically significant difference between these two groups. Also, we found no significant difference in hepatic cholesterol levels between HFD and RD fed uninfected mice (Fig. 2a).

Acute T. cruzi infection significantly alters hepatic lipid levels in mice. a A colorimetric analysis demonstrated a significant increase of cholesterol in the livers of infected mice (n = 5; 50% increase) on either a RD or HFD at day 30 pi. b Magnetic resonance spectroscopy analysis of the livers demonstrated a significant reduction in the levels of lipid (triglyceride)/water ratio during acute T. cruzi infection at day 30 pi. Feeding an HFD increased hepatic lipid (triglyceride) to water ratio (61-fold) compared to mice fed on RD (uninfected group). Infection decreased the levels of lipid to water ratio in both RD and HFD fed mice. The reduction was greater in mice fed on HFD (26.3-fold decrease normalized with uninfected HFD fed mice) compared to RD (12.6-fold decrease normalized with uninfected RD fed mice) during infection. p ≤ 0.005, represented by black stars (n = 5). Bars represent mean values of the data with standard error of the mean (SEM) as vertical lines
Acute T. cruzi infection causes a reduction in the levels of hepatic neutral lipids
We measured hepatic neutral lipid levels (triglycerides) by magnetic resonance spectroscopy (MRS). The lipid to water ratio was measured by MRS in both HFD and RD fed infected mice and compared to the respective uninfected mice. For uninfected mice, the mice fed on HFD demonstrated a significant increase (61-fold) in lipid to water ratio compared to RD fed mice, reflecting an increase in hepatic neutral lipids caused by HFD. T. cruzi infection caused a reduction in the lipid to water ratio in both HFD and RD groups; however, this reduction was significantly greater in mice fed a HFD (26.3- vs 12.6-fold; Fig. 2b).
Acute infection alters liver morphology and physiology
Photomicrographs of H&E-stained liver sections demonstrated significantly altered liver morphology during T. cruzi infection both in RD and HFD fed mice (Fig. 3). In the absence of infection, mice fed on HFD showed increased levels of both macro- and micro-vesicular lipid droplets in liver sections compared to RD fed mice (Supplemental Fig. 2). T. cruzi infection significantly reduced the levels of macro- and micro-vesicular lipid droplets in HFD fed mice compared to HFD fed uninfected mice. Also, micro-vesicular lipid droplets were approximately four times smaller in infected HFD mice than in uninfected HFD fed mice. Lipid droplets were only observed in the hepatic central and portal venules and veins of HFD fed infected mice at day 30 pi. Micro-vesicular lipid droplets were also significantly reduced in the infected RD fed infected mice compared to uninfected RD mice (Supplemental Fig. 2). We also observed increased levels of liver-specific phagocytic Kupffer cells in the sinusoidal space in infected mice fed on either diet (Fig. 3). Immunohistochemical (IHC) analysis of the liver sections with F4/80 staining (specific to Kupffer and macrophages) demonstrated a significant increase in the levels of Kupffer cells and macrophages during acute infection (Supplemental Fig. 3). The levels of inflammatory cells were significantly lowered in the infected mice fed on HFD (50% decrease) compared to RD.

Histological analysis of the livers of acute T. cruzi-infected mice demonstrated inflammation and alteration in the levels of lipid droplets. The photomicrographs of the H&E-stained liver sections demonstrated the presence and increased levels of phagocytic Kupffer cells in the sinusoidal space of the livers in the infected mice compared to uninfected mice irrespective of the diet fed. Acute infection increased the infiltration of inflammatory cells (black arrowhead) especially around the veins in both RD and HFD fed mice as demonstrated by H&E staining. The number of migrated inflammatory cells were lower (40%) in HFD fed uninfected mice compared to RD fed mice at day 30 pi. Liver of mice fed on HFD demonstrated increased levels of lipid droplets both macro (red arrowheads up to 25 μm) and micro (up 5 μm to red arrows) compared to RD fed uninfected mice. Intracellular lipid droplets appeared as uncolored circles in the liver tissues. Infection significantly decreased the levels of lipid droplets in the livers of both RD and HFD fed mice (Supplemental Fig. 2). In the infected HFD fed mice macro-vesicular lipid droplets reduced their size compared to uninfected HFD fed mice (enlarged inset pictures). Also, the livers of HFD fed infected mice displayed lipid droplets (green arrowhead) in the hepatic and portal venules (PV). Five mice were analyzed in these experiments, and representative images are shown
T. cruzi acute infection regulates lipid metabolism in the liver
qPCR analysis of mouse livers was performed to measure the mRNA levels of genes encoding apolipoproteins (Apo) as well as genes regulating cholesterol and fatty acid metabolism and cholesterol efflux. mRNA levels of ApoA, ApoB, and ApoE were significantly reduced compared to uninfected RD fed mice (−1.5- to −5-fold; Fig. 4a). mRNA levels of both ApoB and ApoE were also significantly reduced in the HFD fed infected mice compared to RD fed infected mice. mRNA levels of genes involved in cholesterol biosynthesis such as HMG-CoA reductase (HMGCR), HMG-CoA synthase (HMGCS), and insulin-induced gene 1 (Insig1) were significantly reduced in HFD fed infected mice (4- to 5-fold) compared to RD fed uninfected mice. mRNA levels of fatty acid metabolism genes such as acetyl-CoA acyltransferase 1A (Acaa1a), acyl-CoA dehydrogenase 9 and 10 (Acad9 and Acad10), acyl-CoA oxidase 1 (Acox1), and fatty acid binding protein (Fabp1) were significantly reduced during infection (Fig. 4b). qPCR analysis also demonstrated a significant increase in the mRNA levels of ATP-binding cassette a1 gene (Abca1), which is mainly involved in cholesterol efflux, in infected mice compared to uninfected RD fed mice. Abca1 mRNA levels were also significantly higher in HFD fed infected mice (3560 ± 432-fold) compared to an RD fed infected mice (9.1 ± 1.3-fold) (Fig. 5b). To ask whether this mRNA increase correlated with protein levels, we performed immunoblot analysis of Abca1 (Fig. 5a). Indeed, Abca1 protein levels were significantly higher in the HFD fed infected mice compared to all other experimental groups.

Gene expression changes in the liver of T. cruzi-infected mice fed on regular (RD) or high-fat diet (HFD) indicate alterations in lipid metabolism, inflammation, and oxidative stress. a qPCR analysis demonstrated a significant alterations in the expressions of the mRNA levels of apolipoproteins during acute infection. The mRNA levels of ApoB and ApoE are significantly reduced in the infected HFD fed mice compared to the infected RD fed mice. b mRNA levels of the genes involved in cholesterol mechanism such as HMG-CoA reductase (HMGCR), HMG-CoA synthase (HMGCS), and insulin-induced gene (Insig1) were significantly decreased in the infected HFD fed mice compared to the infected RD fed mice. qPCR analysis showed a significant decrease in the levels of the genes involved in lipid metabolism such as leptin (lep), acetyl-coenzyme A acyltransferase 1A (ACAA1a), Acyl-CoA dehydrogenase (Acad 9), and fatty acid-binding proteins (FABPs) in the livers of the infected HFD fed mice compared to the infected RD fed mice. c qPCR analysis demonstrated a significant increase in the mRNA levels of the genes involved in inflammation such as chemokine (C-C motif) ligand 5 (CCL5), C-C motif receptors (CCR1 and 2), TNF-α, and IFN-γ during acute infection in the livers compared to uninfected mice. The levels of IFN-γ was significantly higher compared to TNF levels. d mRNA analysis validated a significant decrease in the levels of the antioxidant genes such as glutathione peroxidase (GPX 2), EH domain containing 2 (EHD2), peroxiredoxin1(PRDX1), catalase (CAT), superoxide dismutase (SOD), nitrogen synthase (NOS2), and NADPH oxidase 1 (NOX1) during acute infection in the livers compared to uninfected mice. Prostaglandin-endoperoxide synthase 2 (PTGS2) levels were significantly downregulated in the livers of the infected HFD fed mice compared to the infected RD fed mice

Upregulation of cholesterol efflux regulatory protein ATP-binding cassette (ABCA1) in the livers during acute T. cruzi infection. a Immunoblot analysis demonstrated a significant increase in the protein levels of ABCA1 in the livers of acute infected mice compared to uninfected mice fed on either a RD or HFD (n = 5). The levels of ABCA1 protein are significantly higher in the infected HFD fed mice compared to the infected RD fed mice (p ≤ 0.005). The intensities of the bands were measured using ImageJ program and presented as a bar graph and normalized to the levels of GDI protein (AU arbitrary units). b qPCR analysis showed a significant increase in the mRNA levels of ABCA1 in the livers compared to uninfected mice fed on their respective diets at day 30 pi (n = 5). ABCA1 mRNA levels were significantly higher in the infected HFD fed mice compared to the infected RD fed mice
Acute infection induces inflammation and oxidative stress in liver
qPCR analysis demonstrated a significant increase in the mRNA levels of several inflammatory genes such as CCl5, IFN-γ, Itgβ2, Tgfβ, and TNF-α in the livers of infected mice compared to uninfected RD fed mice (Fig. 4c). Interestingly, immunoblot analysis demonstrated a significant increase in the protein levels of IFN-γ in the livers of both RD and HFD fed mice during acute infection (Fig. 6a, b). However, the levels of TNF-α significantly increased only in infected RD fed mice but were significantly decreased in infected HFD mice compared to uninfected mice (Fig. 6a, c).

Acute T. cruzi infection (day 30 pi) regulates inflammation and oxidative stress in the livers (n = 5). a Immunoblot analysis demonstrated a significant change in the protein levels of IFN-γ (b), TNF-α (c), and SOD (d) normalized to the levels of GDI protein. The intensities of the bands were measured using ImageJ program and presented as bar graphs for IFN-γ (b), TNF-α (c), and SOD (d) protein levels (AU arbitrary units). Between the uninfected groups, high-fat diet decreased the levels of IFN-γ, increased TNF-α levels, and showed no significant change in SOD levels in the livers compared to uninfected RD fed mice. b IFN-γ significantly increased during acute infection in both RD and HFD fed infected mice compared to uninfected mice (p ≤ 0.005). c TNF-α significantly increased in RD fed infected mice and decreased in HFD fed infected mice compared to their respective diet fed uninfected mice (p ≤ 0.005). d SOD levels were significantly higher during acute infection at day 30 pi. The protein level of SOD was significantly greater in the infected RD fed mice compared to the infected HFD fed mice (p ≤ 0.005)
qPCR analysis demonstrated a significant reduction in the mRNA levels of genes encoding GPX2, Ehd2, Prdx1, catalase, SOD, Ptgs2, Nos2, and Nox1 in the livers during acute infection irrespective of the diets fed (Fig. 4d). The mRNA levels of antioxidant enzymes such as catalase and superoxide dismutase (SOD) were significantly reduced in the infected HFD fed mice compared to the infected RD fed mice (Fig. 4d). Immunoblot analysis also demonstrated a significantly lower level of SOD, an antioxidant enzyme in the livers of HFD fed infected mice compared to RD fed mice (Fig. 6a, d). These data suggest that oxidative stress is more prominent in the livers of HFD fed infected mice compared to RD fed infected mice (Figs. 4d and 6a, d).
Altered energy metabolism during acute infection
Protein levels of the critical regulators of lipid metabolism such as FAS, UCP3, and AMPK were measured to analyze the effect of diet on the hepatic lipid homeostasis during acute infection. Immunoblot analysis demonstrated a significant reduction in FAS, a key enzyme involved in liver lipid homeostasis (J ensen_Urstad and Semenkovich 2012; Jump 2011), during acute infection (Fig. 7a, b). RD fed infected mice showed a 1.5-fold reduction and HFD fed infected mice showed a 2-fold decrease in the levels of FAS compared to the respective uninfected mice. Among infected mice, FAS levels were also significantly reduced (2-fold) in mice fed a HFD compared to those fed a RD (Fig. 7b). We have also measured the levels of mitochondrial uncoupling protein (UCP3) because fatty acids regulate UCP3 levels (Fig. 7a) (Schrauwen 2004). Indeed, immunoblot analysis demonstrated a significant reduction of UCP3 levels during acute infection in the livers of infected mice fed on either a RD or a HFD (26- and 1.5-fold, respectively) relative to their respective uninfected mice (Fig. 7c). The level of UCP3 in mice fed a HFD was significantly increased (2.5-fold) compared to the mice fed a RD in T. cruzi-infected mice at day 30 pi. AMP-activated protein kinase (AMPK) signaling was analyzed in the livers of infected mice fed on either an HFD or RD during acute infection to evaluate the levels of nutritional stress. Immunoblot analysis demonstrated a significant increase in the levels of pAMPK in the infected RD fed mice and a decrease in the levels of pAMPK in the infected HFD mice compared to their respective uninfected mice (Fig. 7d, e). Furthermore, levels of pMTOR, a downstream regulator of AMPK signaling, decreased in the infected RD fed mice and showed no significant difference in the infected HFD fed mice compared to their respective uninfected mice (Fig. 7d, e).

T. cruzi acute infection alters systemic energy homeostasis of the livers and induces autophagy differently between RD and HFD fed mice. a Immunoblot analyses demonstrated a significant alteration in the levels of fatty acid synthase (FAS) and UCP3 during acute infection. The intensities of the bands were measured using ImageJ program, normalized to the levels of GDI protein (AU arbitrary units), and the representative bar graphs are presented in b and c. b The protein levels of FAS were significantly decreased in both the RD and HFD fed infected mice compared to the uninfected mice. However, the level of FAS was significantly lowered in the infected HFD fed mice compared to the infected RD fed mice (p ≤ 0.005). c The levels of a mitochondrial b-oxidation marker UCP3 were significantly decreased during acute infection. Immunoblot analysis showed a significant reduction in the level of UCP3 in the uninfected HFD fed mice compared to the uninfected RD fed mice (p ≤ 0.005). d Immunoblot analyses demonstrated a significant alteration in the levels of proteins involved in AMPK signaling and autophagy during acute infection. The intensities of the bands were measured using ImageJ program, normalized to the levels of GDI protein (AU arbitrary units), and the representative bar graphs are presented in b and c. e Immunoblot analysis of the livers demonstrated a significant difference in the AMPK signaling between RD and HFD fed and infected and uninfected mice. pAMPK levels were significantly increased in the infected RD fed mice and significantly decreased in the infected HFD fed mice compared to their respective diet fed uninfected mice. pMTOR levels were significantly reduced in the infected RD fed mice and showed no significant change in the infected HFD fed mice compared to their respective diet fed uninfected mice (p ≤ 0.005). f Immunoblot analysis demonstrated a significant increase and decrease in the levels of autophagy markers LC3 and P62, respectively, in the livers of mice fed on either RD or HFD at day 30 pi (p ≤ 0.005)
Acute infection induces autophagy in liver
Autophagy is influenced by nutrient sources and the host metabolic status (Kim et al. 2011; Galluzzi et al. 2014). During nutrient deprivation, autophagosome formation is dramatically induced (Galluzzi et al. 2014). Autophagy stimulated by AMPK activation and mTOR downregulation results in elevated ATP production via recycling of nutrients (Kim et al. 2011). Because we saw a significant increase in pAMPK levels and a downregulation of mTOR in RD fed infected mice and a significant decrease in pAMPK in HFD fed infected mice, we expected to see increased levels of autophagy marker LC3II only in RD fed infected mice. However, immunoblot analysis demonstrated increased levels of LC3II in both RD and HFD fed infected mice (2.2- and 4.9-fold, respectively) compared to their respective uninfected mice (Fig. 7d, f). Interestingly, LC3II levels were also significantly higher in infected HFD fed mice (2.1-fold) compared to infected RD fed mice (Fig. 7d, f). To confirm the upregulation of autophagy during infection, we performed immunoblot analysis of another autophagy marker p62/A170/SQSTM1 (hereafter referred to as “p62”), which is one of the substrates that are specifically degraded through the autophagy–lysosomal pathway (Bjørkøy et al. 2009). The levels of p62 were significantly reduced in the infected livers irrespective of the diets fed (Fig. 7d, f). These data suggest that the autophagy signaling pathway induced during T. cruzi infection is regulated differentially between RD and HFD fed mice. For instance, in RD fed infected mice AMPK/mTOR signaling in response to nutrient deprivation may induce autophagy. However, in HFD fed mice, it is likely that oxidative stress but not AMPK signaling may induce autophagy.
It has been demonstrated that HFD can increase mitochondrial oxidative stress in mice (Matsuzawa-Nagata et al. 2008) and that oxidative stress or increased production of reactive oxygen species can induce autophagy (Navarro-Yepes et al. 2014). Our data demonstrate that loss of hepatic triglyceride levels were greater in HFD fed mice compared to RD fed mice during infection (Fig. 2), suggesting that increased triglyceride catabolism in the livers of HFD fed infected mice may cause enhanced reactive oxygen species (ROS) production and oxidative stress. SOD acts as a first line of defense against ROS. However, we found that the levels of antioxidant enzymes including SOD are significantly reduced in HFD fed infected mice compared to RD fed mice (Fig. 6d). We also observed that the levels of SOD in the livers of RD fed infected mice are 5-fold greater than the uninfected RD fed mice (and 3-fold greater than HFD fed infected mice), which suggests that RD fed infected mice are under less oxidative stress compared to HFD fed infected mice.
Together, these data suggest that diet and T. cruzi infection significantly alter hepatic energy homeostasis, markers of metabolism, inflammation, and oxidative stress.
Discussion
Previously, we demonstrated that feeding a HFD to mice during T. cruzi acute infection decreases the parasite load in the heart but increases it in adipose tissue compared to RD fed mice (Nagajyothi et al. 2014). In the current study, we demonstrated a significantly increased parasite load in the livers of HFD fed mice (Fig. 1). We hypothesize that this increase may be due to the high affinity of T. cruzi for lipids and lipoproteins, because the liver is a major site of lipid biosynthesis, storage, and secretion, and because it functions in lipoprotein metabolism, in particular by absorbing circulatory lipoproteins (Nguyen et al. 2008). Thus, as HFD increases circulatory lipid and lipoprotein levels, this in turn increases their levels in the liver, attracting T. cruzi to the liver and causing an increase in the liver parasite load compared to RD during acute infection.
A normal mouse liver is largely comprised of neutral lipids (e.g., triglycerides, 80%) (Nelson 1962). We analyzed the effect of HFD on the lipid levels in the livers of infected mice and compared to mice fed on RD. MRS data demonstrated a reduction in hepatic triglycerides during infection. This reduction was significantly greater in mice fed a HFD (26.3- vs 12.6-fold; Fig. 2b). Morphological analysis of lipid droplets in the livers of infected mice correlated with the MRS data showing a significant decrease in the lipid/water ratio in HFD infected mice (Fig. 2b and Supplemental Fig. 2). This result suggests that the demand for lipid catabolism in the livers of HFD fed mice is greater than in RD fed mice during acute infection (day 30 pi), which may be due to the higher parasite load in HFD fed mice (Fig. 1).
About 20% of the daily cholesterol is synthesized in the liver. Previously, we showed that T. cruzi has high affinity for lipoproteins and that acute infection results in tissue cholesterol accumulation (Nagajyothi et al. 2011). Here, we showed that hepatic cholesterol accumulation is significant during acute infection in the mice fed either a RD or HFD compared to uninfected mice (Fig. 2a). These data suggest that acute infection induces cholesterol steatosis and triglyceride insufficiency in the liver, which in turn may affect liver function and lipid homeostasis. Indeed, the qPCR analysis data of mouse livers support our lipid analysis data, which was performed to evaluate the altered RNA levels of the liver lipid metabolic proteins. In particular, we measured the mRNA levels of genes encoding Apo as well as genes regulating cholesterol and fatty acid metabolism and cholesterol efflux. The levels of these mRNAs in the livers of infected mice (both RD and HFD groups) were significantly altered compared to uninfected RD fed mice. These data suggest that cholesterol biosynthesis and fatty acid catabolism are significantly altered during acute infection, especially in the HFD fed mice (Figs. 4b and 5). Thus, it is likely that the altered lipid metabolism in the liver contributes to the overall infection progression and affects systemic energy homeostasis.
Infection progression and regression depends on the immune cell response and inflammation signaling of the host. Acute infection results in increased levels of infiltrating inflammatory cells (Nagajyothi et al. 2014). Because we and others previously showed that inflammatory cells infiltrate the hearts during T. cruzi infection (Nagajyothi et al. 2014; Nagajyothi et al. 2012; Gupta et al. 2014; Cunha-Neto et al. 1998), we measured the levels of inflammatory cells in mouse livers. We found that the livers of HFD fed infected mice contained fewer inflammatory cells (50%) compared to RD fed infected mice (Fig. 3 and Supplemental Fig. 3), suggesting that HFD prevents the infiltration of inflammatory cells into the liver during acute infection. Further investigation is required to understand the molecular mechanisms involved in the prevention of hepatic infiltration of inflammatory cells during acute T. cruzi infection in a murine model fed on HFD, as the inflammatory cells play a major role in the pathogenesis of chagasic cardiomyopathy.
The inflammatory response is induced by the parasite load during acute T. cruzi infection (Teixeira et al. 2002). Indeed, our qPCR analysis demonstrated a significant increase in the mRNA levels of several inflammatory genes such as CCl5, IFN-γ, Itgβ2, Tgfβ, and TNF-α in the livers of infected mice compared to uninfected RD fed mice (Fig. 4). The protein levels of TNF-α in infected RD fed mice were significantly higher compared to hepatic TNF-α levels of infected HFD fed mice, which is attributed to the increased levels of F480-positive cells in the livers of infected RD fed mice. However, the levels of IFN-γ significantly increased in the livers of both RD and HFD fed mice during acute infection (Fig. 6a, b). Because it has been shown that IFN-γ attenuates lipid storage in human adipocytes (McGillicuddy et al. 2009), increased IFN-γ may contribute to the loss of triglycerides in the liver during acute infection. Thus, increased levels of IFN-γ may disrupt energy expenditure and metabolic homeostasis (McGillicuddy et al. 2009). IFN-γ is shown to stimulate TNF-α release from the infected macrophages. HFD fed mice showed decreased levels of hepatic macrophages and thus lower levels of TNF-α could contribute to the higher parasite load compared to the infected RD fed mice.
Increased oxidative stress has been demonstrated in several organs/tissues during acute Chagas infection (Wen et al. 2014; Gupta et al. 2009). Indeed, we detected altered mRNA levels of several genes involved in oxidative stress in the livers of infected mice fed on either an RD or HFD. mRNA levels of genes encoding GPX2, Ehd2, Prdx1, catalase, SOD, Ptgs2, Nos2, and Nox1 were significantly reduced during acute infection irrespective of the diets fed (Fig. 4d). Furthermore, the mRNA levels of antioxidant enzymes such as catalase and SOD were significantly reduced in the infected HFD fed mice compared to the infected RD fed mice (Fig. 4d). These data suggest that oxidative stress is more prominent in the livers of HFD fed infected mice compared to RD fed infected mice (Figs. 4d and 6). HFD-induced oxidative stress may contribute to the pathogenesis of chagasic cardiomyopathy.
To understand how diet regulates liver energy homeostasis via lipid metabolism during acute infection, we measured the expression levels of FAS, UCP3, and AMPK, crucial regulators of lipid metabolism and energy homeostasis in the liver (Chakravarthy et al. 2007; Busiello et al. 2015; Hardie et al. 2012). Immunoblot analysis demonstrated a significant reduction in FAS during acute infection in both RD and HFD fed mice compared to the uninfected mice (Fig. 7a, b). This reduction in FAS levels during infection is expected to contribute to a further decrease in triglyceride levels in the livers of infected mice, as well as to nutritional stress (Jensen_Urstad and Semenkovich 2012). Even though the level of UCP3 is significantly decreased during acute infection, HFD fed infected mice showed 2.5-fold higher level of UCP3 compared to RD fed infected mice, suggesting that HFD might have increased the levels of mitochondrial fatty acids during infection. AMP-activated protein kinase (AMPK) plays a key role as a master regulator of cellular energy homeostasis, and its levels and activity depend on the nutritional status (Hardie et al. 2012). Thus, AMPK signaling was analyzed in the livers of infected mice fed on either an HFD or RD during acute infection to evaluate the levels of nutritional stress. Immunoblot analysis demonstrated a significant increase in the levels of pAMPK signaling (reduced pMTOR) in the infected RD fed mice and a decrease in the levels of pAMPK in the infected HFD mice compared to their respective uninfected mice (Fig. 7d, e). As a cellular energy sensor responding to low ATP levels, AMPK activation positively regulates signaling pathways that replenish cellular ATP supplies, including fatty acid oxidation and autophagy (O’Neill et al. 2013; Kim et al. 2011). This suggests that during acute infection RD fed mice are under greater nutritional requirement (i.e., starvation) compared to HFD mice.
Altered lipid metabolism during acute infection regulate AMPK signaling and autophagy. Autophagy is influenced by nutrient sources and the host metabolic status (Kim et al. 2011; Galluzzi et al. 2014). During nutrient deprivation, autophagosome formation is dramatically induced (Galluzzi et al. 2014). Autophagy stimulated by AMPK activation and mTOR downregulation results in elevated ATP production via recycling of nutrients (Kim et al. 2011). Because we saw a significant increase in pAMPK levels and a downregulation of mTOR in RD fed infected mice but a significant decrease in pAMPK in HFD fed infected mice, we expected to see increased levels of autophagy marker LC3II only in RD fed infected mice. However, immunoblot analysis demonstrated increased levels of LC3II in both RD and HFD fed infected mice (2.2- and 4.9-fold, respectively) compared to their respective uninfected mice (Fig. 7d, f). Interestingly, LC3II levels were also significantly higher in infected HFD fed mice (2.1-fold) compared to infected RD fed mice (Fig. 7d, f). To confirm the upregulation of autophagy during infection, we performed immunoblot analysis of another autophagy marker p62/A170/SQSTM1 (hereafter referred to as “p62”), which is one of the specific substrates that are degraded through the autophagy–lysosomal pathway (Bjørkøy et al. 2009). The levels of p62 was significantly reduced in the infected livers irrespective of the diets fed (Fig. 7d, f). These data suggest that the autophagy signaling pathway induced during T. cruzi infection is regulated differentially between RD and HFD fed mice. For instance, in RD fed infected mice AMPK/mTOR signaling in response to nutrient deprivation may induce autophagy. In HFD fed mice, it is likely that oxidative stress and not the regular AMPK signaling may induce autophagy. It has been demonstrated that oxidative stress or increased production of ROS also induces autophagy (Navarro-Yepes et al. 2014). Furthermore, HFD has been shown to increase mitochondrial oxidative stress in mice (Matsuzawa-Nagata et al. 2008). Our data demonstrate that loss of hepatic triglyceride levels were greater in HFD fed mice compared to RD fed mice during infection (Fig. 2b). This suggests that increased triglyceride catabolism in the livers of HFD fed infected mice, which could cause enhanced ROS production and oxidative stress. SOD acts as a first line of defense against ROS. We have demonstrated that the levels of antioxidant enzymes including SOD are significantly reduced in HFD fed infected mice compared to RD fed mice (Fig. 6d). We observed that the levels of SOD in the livers of RD fed infected mice are 5-fold greater than the uninfected RD fed mice (and 3-fold greater than HFD fed infected mice), which suggests that RD fed infected mice are under less oxidative stress compared to HFD fed infected mice.
Together, these observations indicate that the acute T. cruzi infection causes liver inflammation, oxidative stress and loss in lipid droplets levels, all of which were ameliorated by HFD which may alter liver energy metabolism.
Conclusion
The liver is a key metabolic organ governing body energy metabolism. It acts as a central hub to metabolically connect to various tissues, including the heart and adipose tissue. It has been shown that T. cruzi infection causes enlargement of the liver and that the parasites persist in the liver during infection (Sardinha et al. 2010; Collier et al. 1942). It has also been demonstrated that feeding CD1 mice acutely infected with T. cruzi a HFD increases their survival (Nagajyothi et al. 2014; Brima et al. 2015). Here, we report that although HFD decreases the parasite load in the hearts of acutely infected CD1 mice, it increases the hepatic parasite load compared to the mice fed on RD. We also found that acute T. cruzi infection alters energy and metabolic homeostasis of the liver. Furthermore, the livers of mice fed on a HFD showed significantly altered lipid homeostasis compared to RD fed mice. We also found that acute infection caused severe loss of triglycerides in the livers of infected mice irrespective of the diets (HFD or RD). Our data also indicate that the livers of infected mice use autophagy induced by two different mechanisms, which depend on the diet type, to overcome nutritional and oxidative stress during acute infection. In particular, RD fed infected mice upregulate AMPK/mTOR/autophagy signaling pathway to overcome energy deprivation induced by T. cruzi infection. Autophagy induced during acute infection may deliver lipid droplets (lipophagy) as a source of energy and prevent nutritional stress-induced cell death. HFD fed infected mice also utilize autophagy as a regulator of oxidative stress to prevent ROS induced cell death.
Autophagy plays a pivotal role not only in normal liver physiology but also in the pathogenesis of many liver diseases including nonalcoholic and alcoholic fatty liver and viral hepatitis (Mao et al. 2016; Tang et al. 2012). Acute T. cruzi infection may adapt lipophagic pathway to survive energy starvation by increasing autophagic sequestration of lipid droplets and their degradation in hepatic lysosomes. Further work is needed to understand the mechanisms which interlink lipophagy to autophagy during acute and chronic infection. We conclude that T. cruzi infection alters hepatic lipid and energy homeostasis and that HFD directly contributes to the alterations in lipid metabolism, inflammation, and oxidative stress during acute infection in CD1 mice. Further studies in a chronic chagasic model fed on different diets may establish a link between liver lipid metabolism and the pathogenesis of chagasic cardiomyopathy, which is of a greater importance to chagasic patients to improve lipid metabolism. In sum, our results indicate that autophagy is induced in the livers of acutely infected mice as a survival mechanism, which is differentially regulated by diet.
References
Bern C, Martin DL, Gilman RH (2011) Acute and congenital Chagas disease. Adv Parasitol 75:19–47. doi:10.1016/B978-0-12-385863-4
Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T (2009) Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 452:181–197. doi:10.1016/S0076-6879(08)03612-4
Bonney KM (2014) Chagas disease in the 21st century: a public health success or an emerging threat? Parasite 21:1–10. doi:10.1051/parasite/2014012
Brima W1, Eden DJ, Mehdi SF, Bravo M, Wiese MM, Stein J, Almonte V, Zhao D, Kurland I, Pessin JE, Zima T, Tanowitz HB, Weiss LM, Roth J, Nagajyothi F (2015) The brighter (and evolutionarily older) face of the metabolic syndrome: evidence from Trypanosoma cruzi infection in CD-1 mice. Diabetes Metab Res Rev 31:346–359. doi:10.1002/dmrr.2636
Busiello RA, Savarese S, Lombardi A (2015) Mitochondrial uncoupling proteins and energy metabolism. Front Physiol 6:1–7. doi:10.3389/fphys.2015.00036
Chakravarthy MV, Zhu Y, López M, Yin L, Wozniak DF, Coleman T, Hu Z, Wolfgang M, Vidal-Puig A, Lane MD, Semenkovich CF (2007) Brain fatty acid synthase activates PPARα to maintain energy homeostasis. J Clin Invest 117:2539–2552. doi:10.1172/JCI31183
Chu KY, O’Reilly L, Ramm G, Biden TJ (2015) High-fat diet increases autophagic flux in pancreatic beta cells in vivo and ex vivo in mice. Diabetologia 58:2074–2078. doi:10.1007/s00125-015-3665-x
Collier HO, Fulton JD, Innes JR (1942) The oedema of mice infected with Trypanosoma cruzi, and the accompanying pathological lesions. Ann Trop Med Parasitol 36:137–150. doi:10.1080/00034983.1942.11685149
Combs TP, Nagajyothi F, Mukherjee S, de Almeida CJ, Jelicks LA, Schubert W, Lin Y, Jayabalan DS, Zhao D, Braunstein VL, Landskroner-Eiger S, Cordero A, Factor SM, Weiss LM, Lisanti MP, Tanowitz HB, Scherer PE (2005) The adipocyte as an important target cell for Trypanosoma cruzi infection. J Biol Chem 280:24085–24094. doi:10.1074/jbc.M412802200
Cunha-Neto E, Rizzo LV, Albuquerque F, Abel L, Guilherme L, Bocchi E, Bacal F, Carrara D, Ianni B, Mady C, Kalil J (1998) Cytokine production profile of heart-infiltrating T cells in Chagas disease cardiomyopathy. Braz J Med Biol Res 1:133–137. doi:10.1590/S0100-879X1998000100018
Galluzzi L, Pietrocola F, Levine B, Kroemer G (2014) Metabolic control of autophagy. Cell 159:1263–1276. doi:10.1016/j.cell.2014.11.006
Grayson M (2010) Chagas disease. Nature 465:S3. doi:10.1038/465S3a
Gupta S, Wen JJ, Garg NJ (2009) Oxidative stress in Chagas disease. Interdiscip Perspect Infect Dis 2009:190354. doi:10.1155/2009/190354
Gupta S1, Silva TS, Osizugbo JE, Tucker L, Spratt HM, Garg NJ (2014) Serum-mediated activation of macrophages reflects TcVac2 vaccine efficacy against chagas disease. Infect Immun 82:1382–1389. doi:10.1128/IAI.01186-13
Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13:251–262. doi:10.1038/nrm3311
Hsu HC, Chen CY, Lee BC, Chen MF (2015) High-fat diet induces cardiomyocyte apoptosis via the inhibition of autophagy. Eur J Nutr. doi:10.1007/s00394-015-1034-7
Jensen-Urstad AP, Semenkovich CF (2012) Fatty acid synthase and liver triglyceride metabolism: housekeeper or messenger? Biochim Biophys Acta 1821:747–753. doi:10.1016/j.bbalip.2011.09. 017
Johndrow C, Nelson R, Tanowitz H, Weiss LM, Nagajyothi F (2014) Trypanosoma cruzi infection results in an increase in intracellular cholesterol. Microbes Infect 16:337–344. doi:10.1016/j.micinf.2014.01.001
Jump DB (2011) Fatty acid regulation of hepatic lipid metabolism. Curr Opin Clin Nutr Metab Care 14:115–120. doi:10.1097/MCO.0b013e328342991c
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141. doi:10.1038/ncb2152
Liu Y, Palanivel R, Rai E, Park M, Gabor TV, Scheid MP, Xu A, Sweeney G (2015) Adiponectin stimulates autophagy and reduces oxidative stress to enhance insulin sensitivity during high-fat diet feeding in mice. Diabetes 64:36–48. doi:10.2337/db14-0267
Mao Y, Yu F, Wang J, Guo C, Fan X (2016) Autophagy: a new target for nonalcoholic fatty liver disease therapy. Hepat Med 8:27–37. doi:10.2147/HMER.S98120
Matsuzawa-Nagata N, Takamura T, Ando H, Nakamura S, Kurita S, Misu H, Ota T, Yokoyama M, Honda M, Miyamoto K, Kaneko S (2008) Increased oxidative stress precedes the onset of high-fat diet–induced insulin resistance and obesity. Metabolism 57:1071–1077. doi:10.1016/j.metabol.2008.03.010
McGillicuddy FC, Chiquoine EH, Hinkle CC, Kim RJ, Shah R, Roche HM, Smyth EM, Reilly MP (2009) Interferon γ attenuates insulin signaling, lipid storage and differentiation in human adipocytes via activation of the JAK/STAT pathway. J Biol Chem 284:31936–31944. doi:10.1074/jbc.M109.061655
Nagajyothi F, Machado FS, Burleigh BA, Jelicks LA, Scherer PE, Mukherjee S, Lisanti MP, Weiss LM, Garg NJ, Tanowitz HB (2012) Mechanisms of Trypanosoma cruzi persistence in Chagas disease. Cell Microbiol 14:634–643. doi:10.1111/j.1462- 5822.2012.01764
Nagajyothi F, Weiss LM, Silver DL, Desruisseaux MS, Scherer PE, Herz J, Tanowitz HB (2011) Trypanosoma cruzi utilizes the host low density lipoprotein receptor in invasion. PLoS Negl Trop Dis 5:e953. doi:10.1371/journal.pntd.0000953
Nagajyothi F, Weiss LM, Zhao D, Koba W, Jelicks LA, Cui MH, Factor SM, Scherer PE, Tanowitz HB (2014) High fat diet modulates Trypanosoma cruzi infection associated myocarditis. PLoS Negl Trop Dis 8:e3118. doi:10.1371/ journal. pntd .0003118
Navarro-Yepes J, Burns M, Anandhan A, Khalimonchuk O, del Razo LM, Quintanilla-Vega B, Pappa A, Panayiotidis MI, Franco R (2014) Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxid Redox Signal 21:66–85. doi:10.1089/ars.2014.5837
Nelson GJ (1962) The lipid composition of normal mouse liver. J Lipid Research 3:256–262
Nguyen P, Leray V, Diez M, Serisier S, Bloc’h JL, Siliart B, Dumon H (2008) Liver lipid metabolism. J Anim Physiol Anim Nutr 92:272–283. doi:10.1111/j.1439- 0396.2007.00752.x
O’Neill HM1, Holloway GP, Steinberg GR (2013) AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: implications for obesity. Mol Cell Endocrinol 366:135–151. doi:10.1016/j.mce.2012.06.019
Rodriguez-Guardado A, González ML, Rodriguez M, Flores-Chavez M, Boga JA, Gascon J (2015) Trypanosoma cruzi infection in a Spanish liver transplant recipient. Clin Microbiol Infect 21:687e1–687e3. doi:10.1016/j.cmi.2015.03.022
Russell RC, Yuan HX, Guan KL (2014) Autophagy regulation by nutrient signaling. Cell Res 24:42–57. doi:10.1038/cr.2013.166
Sardinha LR, Mosca T, Elias RM, do Nascimento RS, Gonçalves LA, Bucci DZ, Marinho CR, Penha-Gonçalves C, Lima MR, Álvarez JM (2010) The liver plays a major role in clearance and destruction of blood trypomastigotes in Trypanosoma cruzi chronically infected mice. PLoS Negl Trop Dis 4:e578. doi:10.1371/journal.pntd.0000578
Schneider J, Cuervo AM (2014) Liver autophagy: much more than just taking out the trash. Nat Rev Gastroenterol Hepatol 11:187–200. doi:10.1038/nrgastro.2013.211
Schrauwen P, Hesselink MK (2004) The role of uncoupling protein 3 in fatty acid metabolism: protection against lipotoxicity? Proc Nutr Soc 63:287–292. doi:10.1079/ PNS 2004336
Shikanai-Yasuda MA, Carvalho NB (2012) Oral transmission of Chagas disease. Clin Infect Dis 54:845–852. doi:10.1093/cid/cir956
Tang SW, Ducroux A, Jeang KT, Neuveut C (2012) Impact of cellular autophagy on viruses: insights from hepatitis B virus and human retroviruses. J Biomed Sci 19:1–11. doi:10.1186/1423-0127-19-92
Tanowitz HB, Kirchhoff LV, Simon D, Morris SA, Weiss LM, Wittner M (1992) Chagas disease. Clin Microbiol Rev 5:400–419. doi:10.1128/CMR.5.4.400
Tanowitz HB, Weiss LM, Montgomery SP (2011) Chagas disease has now gone global. PLoS Negl Trop Dis 5:e1136. doi:10.1371/journal.pntd.0001136
Teixeira MM, Gazzinelli RT, Silva JS (2002) Chemokines, inflammation and Trypanosoma cruzi infection. Trends Parasitol 18:262–265. doi:10.1016/S1471-4922(02)02283-3
Wen JJ, Nagajyothi F, Machado FS, Weiss LM, Scherer PE, Tanowitz HB, Garg NJ (2014) Markers of oxidative stress in adipose tissue during Trypanosoma cruzi infection. Parasitol Res 113:3159–3165. doi:10.1007/s00436-014-3977-7
Acknowledgements
We acknowledge Dazhi Zhao, Department of Pathology at Albert Einstein College of Medicine, NY, for the technical help. We thank Erika Shor at the Public Health Research Institute for a critical reading of the manuscript. We thank Dr. Joan Durbin, Professor of Pathology at Rutgers New Jersey Medical School for her suggestions on the histology slides. This study was supported by grants from the National Heart, Lung, and Blood Institute (National Institutes of Health HL-112099 and HL-122866) to Jyothi Nagajyothi.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All animal experimental protocols were approved by the Institutional Animal Care and Use Committees (IACUC) of Albert Einstein College of Medicine (No. 20100204).
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Kezia Lizardo, Vanessa Almonte, and Calvin Law contributed equally to this article.
Electronic Supplementary Material
Supplementary Fig. 1
(TIFF 1876 KB)
Supplementary Fig. 2
(TIFF 1342 KB)
Supplementary Fig. 3
(TIFF 6753 KB)
Rights and permissions
About this article

Cite this article
Lizardo, K., Almonte, V., Law, C. et al. Diet regulates liver autophagy differentially in murine acute Trypanosoma cruzi infection. Parasitol Res 116, 711–723 (2017). https://doi.org/10.1007/s00436-016-5337-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-016-5337-2