
Abstract
Although the existence of a sylvatic transmission cycle of Leishmania spp., independent from the domestic cycle, has been proposed, data are scarce on Leishmania infection in wild mammals in Greece. In this study, we aimed to investigate the presence of Leishmania infection in the European brown hare in Greece, to infer the phylogenetic position of the Leishmania parasites detected in hares in Greece, and to identify any possible correlation between Leishmania infection in hares with environmental parameters, using the geographical information system (GIS). Spleen samples from 166 hares were tested by internal transcribed spacer-1 (ITS-1)-nested PCR for the detection of Leishmania DNA. Phylogenetic analysis was performed on Leishmania sequences from hares in Greece in conjunction with Leishmania sequences from dogs in Greece and 46 Leishmania sequences retrieved from GenBank. The Leishmania DNA prevalence in hares was found to be 23.49 % (95 % confidence interval (CI) 17.27–30.69). The phylogenetic analysis confirmed that the Leishmania sequences from hares in Greece belong in the Leishmania donovani complex. The widespread Leishmania infection in hares should be taken into consideration because under specific circumstances, this species can act as a reservoir host. This study suggests that the role of wild animals, including hares, in the epidemiology of Leishmania spp. in Greece deserves further elucidation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Leishmaniasis is a vector-borne mammalian disease caused by a protozoan flagellate of the genus Leishmania, transmitted by phlebotomine sandfly species. Dogs are the major reservoir host of Leishmania infantum. However, Leishmania infection has been also described in cats, equines, and wild mammals including lagomorphs (Gramiccia 2011; Molina et al. 2012; Ruiz-Fons et al. 2013; Moreno et al. 2014; Jiménez et al. 2014; Souza et al. 2014).
Greece is considered to be an endemic country for leishmaniasis. The visceral form of the disease, caused by L. infantum, is the predominant one, endemic in nearly all geographical areas of the country. Cutaneous leishmaniasis, caused by Leishmania tropica and L. infantum in some cases, is also present but it occurs sporadically (Gkolfinopoulou et al. 2013). The overall reported seroprevalence in the canine population in seven regions of the Greek mainland was nearly 20 % ranging from 2.05 % in Florina to 30.12 % in Attiki (Athanasiou et al. 2012), while the seroprevalence was 3.87 % in clinically healthy stray cats living in the area of Thessaloniki (Diakou et al. 2009), and a very low seroprevalence (0.3 %) has been reported in horses in Attiki (Kouam et al. 2010).
Although the existence of a sylvatic transmission cycle that is independent from the domestic cycle has been proposed (Sobrino et al. 2008; Molina et al. 2012; Millán et al. 2014), there is a scarcity of data on Leishmania infection in wild mammals in Greece, a highly endemic country for canine leishmaniasis. Regarding the lagomorphs, there are no data in the countries of the Mediterranean basin including Greece. However, several studies conducted in Spain revealed a 43.6 % Leishmania DNA prevalence (Ruiz-Fons et al. 2013) and 74.1 % seroprevalence in hares (Moreno et al. 2014). Moreover, four out of seven apparently healthy hares submitted to xenodiagnosis were able to infect a mean 4.7 % Phlebotomus perniciosus sandflies (0–10.6 %), a competent L. infantum vector, suggesting that hares may represent a secondary reservoir in the sylvatic transmission cycle of L. infantum in that region (Molina et al. 2012).
Sequence data of the ribosomal RNA (rRNA) gene, in particular the two highly variable internal transcribed spacer regions (ITS-1 and ITS-2), have been used to resolve taxonomic questions and to determine phylogenetic affinities among closely related Leishmania species (Dávila and Momen 2000; Schönian et al. 2000; El Tai et al. 2001; Kuhls et al. 2005; Parvizi et al. 2008; Yang et al. 2010; Wang et al. 2010; Hajjaran et al. 2013). The sequences of seven Leishmania parasites detected in hares were used for the phylogenetic analysis in conjunction with three sequences of Leishmania parasites detected in dogs in Greece and 46 Leishmania sequences retrieved from GenBank. Our work is a first attempt to investigate the presence of Leishmania infection in hares (Lepus europaeus) in Greece, to identify the environmental parameters related to Leishmania infection in hares using geographical information system (GIS) and statistical analysis, and to infer the phylogenetic position of the Leishmania sequences from hares in Greece.
Materials and methods
Hare and canine samples, DNA extraction, ITS-1-nested PCR, and sequencing
Spleen hare samples (n = 166) were collected in the prefectures of Thessaloniki (n = 82) and Chalkidiki (n = 84) (Northern Greece) from 2007 to 2011. The samples were stored at the Research Division, Hunting Federation of Macedonia and Thrace, Thessaloniki, Greece and later submitted still frozen to the Laboratory of Microbiology and Parasitology, Faculty of Veterinary Medicine, University of Thessaly, Greece, where they were stored at −80 °C. Data on hare specimens were located in the field using handheld global positioning system (GPS) units. Canine lymph node aspirates were collected from three dogs with diagnosed canine leishmaniasis in the prefecture of Thessaloniki. The canine samples were submitted to the Laboratory of Microbiology and Parasitology, University of Thessaly, Greece, within 24 h after collection, and they were immediately stored at −20 °C pending DNA extraction.
Total genomic DNA extraction was performed using a commercially available DNA extraction kit (Thermo Scientific GeneJET Genomic DNA Purification Kit) according to the manufacturer’s protocol. The purified DNA was stored at −20 °C.
The samples were analyzed by ITS-1-nested PCR (ITS-1 nPCR) as described previously (Leite et al. 2010). Primers addressed to ITS-1 between the genes coding for SSU rRNA and 5.8SrRNA were used. For the first amplification, 5 μl of DNA solution was added to 45.0 μl of PCR mix containing 25 pmol of the primers 5′-CTGGATCATTTTCCGATG-3′ and 5′-TGATACCACTTATCGCACTT-3′ and 0.2 mM deoxynucleoside triphosphates, 1.5 mM MgCl2, 5 mM KCl, 75 mM Tris–HCl pH 9.0, and 2 U of AmpliTaq DNA polymerase (Applied Biosystems). The cycling conditions were initial denaturation at 95 °C for 2 min followed by 34 cycles consisting of denaturation at 95 °C for 20 s, annealing at 53 °C for 30 s, and extension at 72 °C for 1 min, followed by a final extension at 72 °C for 6 min. Amplification products were subjected to electrophoresis in 2 % agarose gel stained with ethidium bromide (0.5 μg/ml) and visualized under ultraviolet light. The PCR product size stays between 300 and 350 bp. For the second amplification, 10.0 μl of a 1:40 dilution of the first PCR product was added to 15 μl of PCR mix under the same conditions as the first amplification but with the following primers: 5′-CATTTTCCGATGATTACACC-3′ and 5′-CGTTCTTCAACGAAATAGG-3′. Amplification products were visualized on 2 % agarose gel stained with ethidium bromide (0.5 μg/ml). The PCR product size stays between 280 and 330 bp.
Amplicons from seven randomly selected Leishmania PCR-positive hares were sequenced in order to perform the initial identification of the phylogenetic position of the Leishmania parasites detected in hares within the Leishmania spp. Furthermore, amplicons from three Leishmania PCR-positive dogs were sequenced. Sequence analysis was performed on a part of the rRNA gene (ITS-1 region). The positive PCR product was purified using the PureLink PCR purification kit (Invitrogen) and was bidirectionally sequenced using the fluorescent BigDye Terminator Cycle sequencing kit v3.1 (Applied Biosystems), followed by fragment separation with a 3730xl DNA analyzer (Applied Biosystems). The sequences have been deposited in GenBank under accession numbers KP410398–KP410407 (Table 1), and they were used for the phylogenetic analysis in this study.
GIS and statistical analysis
The GIS layers were obtained from climate, elevation, and land cover data bases acquired from the network. Altitude was extracted from a digital elevation model (DEM) with a spatial resolution of 1 km2. Land uses were determined from the Corine Land Cover 2000 database (European Environment Agency (EEA)) and the ArcGIS online application. Climate data were derived from World Clim database.
The livestock raw data were provided from Payment and Control Agency for Guidance and Guarantee Community Aid. New layers were created to represent the towns and villages, the distance from the nearest village, the distance from water presence, and the density of livestock. Crop types were calculated in a buffer zone of 1 km from the sample coordinates (Table 2). All data layers were converted to a common projection, map extent, and resolution. ArcGIS 10.1 GIS software (ESRI, Redlands, CA, USA) was used for data analysis.
Statistical analysis was performed with IBM SPSS 22.0 using basic descriptive and multivariate statistics (Gray and Kinnear 2012). We used the aforementioned bioclimatic and environmental variables in order to test if the bivariate dependent variable (DV)—Leishmania-positive or Leishmania-negative hares—is related to some of the independent variables (IV). Because of the big set of IVs, we first tried to find some factors (new variables) which may represent the most of the variability of the data. We used the exploratory factor analysis for this. The purpose was to use these factors in a bivariate logistic regression model that would be able to predict the positive hares when some IVs would take a range of values (Tabachnick and Fidell 2007). We set the significance level to p = 5 %.
Phylogenetic analysis
Molecular evolutionary analyses were conducted on nucleotide sequences of Leishmania parasites detected in hares and dogs in Greece and on Leishmania sequences that were retrieved from the EMBL database, using the program MEGA 6 (Table 1).
To determine the appropriate model of sequence evolution and statistically compare successively nested more parameter-rich models for this data set, the program MODELTEST Version 3.6 (Posada and Crandall 1998) was used. With a statistical significance of p = 0.01, the HKY85 model (Hasegawa et al. 1985), with gamma correction, obtained the best likelihood score and was thus selected for subsequent analysis. Maximum parsimony (MP) tree was constructed under the heuristic search option with 100 random-taxon-addition replicates and tree bisection–reconnection branch swapping, using PAUP*. Node support was assessed on the basis of 1,000 bootstrap replicates.
A Bayesian analysis was also performed with MRBAYES version 3.1 (Huelsenbeck and Ronquist 2001), under the HKY85 model of sequence evolution. Depending on the data set, random starting trees run for 2 × 106 to 8 × 106 generations were used, sampled every 100 generations. Burn-in frequency was set to the first 25 % of the sampled trees. With some minor differences, the phylogenetic tree based on the Bayesian analysis as well as MP tree showed similar topologies (the tree is available on request).
Results
ITS-1-nested PCR and sequencing
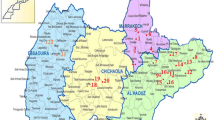
Overall, 39 out of 166 hare samples (23.49 %, 95 % CI 17.27–30.69) were positive for the presence of Leishmania DNA (Thessaloniki 17.1 %, 95 % CI 9.7–27.0; Chalkidiki 29.8 %, 95 % CI 20.3–40.7). The distribution of the Leishmania-positive hares is shown in Fig. 1. The three canine samples collected from dogs diagnosed with canine leishmaniasis were PCR-positive. Seven amplicons from Leishmania PCR-positive hares and the three amplicons from Leishmania PCR-positive dogs were sequenced.

Map of Greece showing the geographical distribution of Leishmania PCR-positive hares between 2007 and 2011 in the prefectures of Thessaloniki and Chalkidiki. Red and blue dots indicate the Leishmania PCR-positive and the Leishmania PCR-negative hares, respectively
GIS and statistical analysis
The Leishmania-positive hares were found in shrubland with pastures, in agricultural areas, and in broadleaved forests with a mean altitude of 225.38 m asl (range 35–750 ± 200.48 SD), while the mean distance from villages and towns was 2.472 m asl (range 850–4,661 ± 1,029.02 SD). Fifty-four percent and 42 % of the positive hare samples were found in crop types of wheat fields and in olive tree plantations, respectively. The remaining 4 % of the positive hare samples were found in cotton and forage plants types. The mean livestock density in the study area (1 km buffer zone) was 38.42 livestock animals (range 0–200 ± 47.36 SD). The number of sheep and goats ranged from 5 to 880 (±151.9 SD), and the number of cattle ranged from 2 to 330 (±77.48 SD). The positive hare samples were detected within a mean distance of 1,169.85 m (range 10–7,285 ± 1,240.94 SD) from bodies of water. The mean annual temperature was 14.8 °C (range 11.7–16.4 ± 12.24 SD) in the prefectures studied during the study period, while the mean annual precipitation recorded was 474.82 mm (range 443–527 ± 23.76 SD) and the annual average humidity reached 67.5 %.
We used the exploratory factor analysis three times: one to reduce the variables related to temperature, one to reduce the variables related to precipitation, and one for the vegetation index variables. Using the criterion of eigenvalues of more than 1, the procedure generated three factors for every group of variables. By entering these variables to the bivariate logistic regression procedure, together with the variables related to the distance from rivers, villages, etc., we did not find a single significant relationship between the DV and the IVs. We then checked the simple biserial correlations between the DV and the IVs. We found that five bioclimatic variables were correlated with the DV, as shown in Table 3. We used these variables in a logistic regression procedure and found only one slightly significant relationship. We found that the presence of Leishmania DNA in hares may be influenced by the precipitation seasonality variable. This variable is produced by the division of the standard deviation of the annual precipitation with the mean value of annual precipitation, that is, the coefficient of variation. However, this relationship is significant only at the 90 % level. Table 3 shows the results of the logistic regression. We can interpret this finding as follows: we are 90 % sure that an increase of one unit in the precipitation seasonality is expected to increase by 6.1 % the odds of hares to be Leishmania positive. This means that in such a case, a hare will have a probability of about 52 % to be Leishmania positive. Since we do not know the real probability for positives (instead of the initial scenario of 50 % probability), the above finding may be indicative. However, the fact that the 95 % confidence interval for the odds includes value 1 should be taken into consideration.
Phylogenetic analysis
The phylogenetic analysis performed on 56 Leishmania sequences, including seven Greek hare and three Greek canine Leishmania sequences, revealed that the homology of the nucleotide sequences between the seven Greek Leishmania sequences from hares was 99.1 %. The homology of the nucleotide sequences between the seven hare and the three canine Leishmania sequences from Greece was 98.9 %, as well as the homology of the nucleotide sequences between the Greek Leishmania sequences and the strains belonging to Leishmania donovani complex which were retrieved from GenBank.
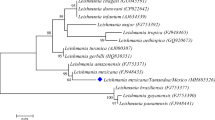
The Greek sequences belonged to the L. donovani complex, and they did not form a monophyletic group. Two major clades (I and II) could be recognized for the L. donovani complex in the maximum parsimony tree based on the ITS-1 sequences (Fig. 2). Clade I included six Leishmania sequences detected in hares in Greece. These sequences formed a strongly supported clade (clade I: Leishmania sp.; BP = 81 %) which was further divided into two subclades (Ia and Ib). Clade II included three canine (GRE1 dog, GRE2 dog, GRE3 dog) and one hare Leishmania sequence from Greece (GRE3 hare) and all the L. donovani complex strains retrieved from GenBank. This clade was further divided into two subclades (IIa and IIb).

Phylogenetic tree constructed based on ITS-1 sequences of 56 Leishmania isolates in this study. Phylogenetic tree based on maximum parsimony analysis from 56 Leishmania sequences (ITS-1 between the genes coding for SSU rRNA and 5.8SrRNA) including the ten Greek Leishmania sequences under study, GRE1 hare, GRE2 hare, GRE3 hare, GRE4 hare, GRE5 hare, GRE6 hare, GRE7 hare, GRE1 dog, GRE2 dog, and GRE3 dog. With some minor differences, the phylogenetic tree based on the Bayesian analysis (available on request) showed similar topologies. The main branches exhibited high bootstrap values and high posterior probabilities (Bayesian analysis) and clearly separated groups in distinct clades
Discussion
This study was a preliminary attempt to detect Leishmania infection in European brown hares (L. europaeus) in Greece and to identify the environmental parameters related to Leishmania infection using GIS and statistical analysis. Moreover, we aimed to infer the phylogenetic position of the Leishmania sequences from hares and dogs in Greece. This is the first report of Leishmania infection in hares in Greece, and our results are in close agreement with the previous reports regarding the susceptibility of hares to Leishmania infection (Ruiz-Fons et al. 2013). The high prevalence of infection (23.49 %) found in our study together with the infectiousness of hares to sandflies and the feeding preference of the latter for hares as documented earlier in Spain (Molina et al. 2012; Jiménez et al. 2013) raises a concern about the role of hares in the epidemiology of Leishmania spp. in Greece. Moreover, the similarity detected between the hare and the canine Leishmania sequences from Greece (98.9 % homology of the nucleotide sequences) could probably be indicative of a possible overlapping of wild and domestic transmission cycles of Leishmania spp. However, this hypothesis needs further investigation.
Interestingly, the GIS analysis revealed that the positive hare samples were found in a mean altitude of 225.38 m above sea level (range 35–750 ± 200.48 SD), while the mean distance was 2.472 m (range 850–4,661 ± 1,029.02 SD) and 1,169.85 m (range 10–7,285 ± 1,240.94 SD) from villages and towns and bodies of water, respectively. Moreover, in a buffer zone of 1 km, the mean livestock density was 38.42 (range 0–200 ± 47.36 SD). The Leishmania-positive hares were found in areas with 67.5 % annual average humidity, 474.82 mm (range 443–527 ± 23.76 SD) mean annual precipitation, and 14.8 °C (range 11.7–16.4 ± 12.24 SD) mean annual temperature. The statistical analysis showed that the presence of Leishmania DNA in hares may be influenced by the precipitation seasonality variable, suggesting that an increase of one unit in the precipitation seasonality is expected to increase the odds of hares to be Leishmania positive by 6.1 %. However, this relationship was significant only at the 90 % level, and it should be correlated with the vector abundance and the phlebotomine sandfly species diversity.
The hare and canine Leishmania sequences in this study had a basal position in the L. donovani complex. Six hare sequences (clade I) formed a separate branch from the three canine and one hare Leishmania sequence from Greece and all the L. donovani complex strains retrieved from GenBank (clade II).
The mean annual incidence of reported human leishmaniasis cases in Greece between 1998 and 2011 was 0.36 cases per 100,000 population, with fluctuation during this period, generally decreasing after 2007, with a small re-increase in 2011 (Gkolfinopoulou et al. 2013). During our study time period, three human cases were reported in the prefecture of Thessaloniki, whereas no human cases were reported in the prefecture of Chalkidiki (HCDCP). Regarding the canine population, the seropositivity reported from 2005 to 2010 was 29.81–42.00 % in Thessaloniki and 0.01–10.66 % in Chalkidiki (Ntais et al. 2013). Thus far, we have no indication that there is an association between the human leishmaniasis cases and the infected hares in the prefectures studied partly due to the low number of officially reported human cases and also probably because they do not share the same living space. Moreover, the Leishmania DNA prevalence found in hares in Chalkidiki and Thessaloniki in conjunction with the seropositivity rates in canine population (Ntais et al. 2013) and the reported human leishmaniasis cases (HCDCP) in the same prefectures during the same study period leads to the hypothesis that leishmaniasis is still in the sylvatic transmission cycle in Chalkidiki, whereas it seems that an overlapping of sylvatic and domestic transmission cycles occurs in Thessaloniki. It is well known that leishmaniasis involves a complex interplay between the protozoon pathogen Leishmania, the arthropod vectors sandflies, the environmental influence on vector distribution, the primary and secondary reservoirs of infection, and the susceptible human populations, thus making necessary the simultaneous investigation of these factors in order to infer the role of a mammalian host in the epidemiology of leishmaniasis. However, the investigation of Leishmania infection in different hosts is crucial in order to design further epidemiological studies, control, and prevention strategies. Thus, the widespread Leishmania infection in hares in the prefectures studied in Greece should be taken into consideration because as reported by Molina et al. (2012), under specific circumstances including an unusual increase in hare population, high density of sandflies, and a low level of immunity in the human population, this species can act as a reservoir host.
Further studies including Leishmania sequences from human cases, domestic and wild animals, and sandflies from Greece and the neighboring countries will give insight into the phylogenetic position of the Leishmania spp. circulating in Greece and the evolutionary history among Greek isolates. The role of the different hosts in the epidemiology of leishmaniasis in Greece deserves further investigation together with the estimation of the vector abundance, distribution, and diversity. In particular, the role of wild mammals in the transmission cycle of Leishmania spp. which remains largely unknown in Greece, a highly endemic country for canine leishmaniasis, needs further elucidation. Studies in endemic and non-endemic areas in the mainland and the islands of Greece, including the vector abundance and the species diversity, will reveal the role of hares in the epidemiology of Leishmania spp. in Greece.
References
Athanasiou LV, Kontos VI, Saridomichelakis MN, Rallis TS, Diakou A (2012) A cross-sectional sero-epidemiological study of canine leishmaniasis in Greek mainland. Acta Trop 122:291–295
Dávila AM, Momen H (2000) Internal-transcribed-spacer (ITS) sequences used to explore phylogenetic relationships within Leishmania. Ann Trop Med Parasitol 94:651–654
Diakou A, Papadopoulos E, Lazarides K (2009) Specific anti-Leishmania spp. antibodies in stray cats in Greece. J Feline Med Surg 11:728–730
El Tai NO, El Fari M, Mauricio I, Miles MA, Oskam L, El Safi SH, Presber WH, Schönian G (2001) Leishmania donovani: intraspecific polymorphisms of Sudanese isolates revealed by PCR-based analyses and DNA sequencing. Exp Parasitol 97:35–44
Gkolfinopoulou K, Bitsolas N, Patrinos S, Veneti L, Marka A, Dougas G, Pervanidou D, Detsis M, Triantafillou E, Georgakopoulou T, Billinis C, Kremastinou J, Hadjichristodoulou C (2013) Epidemiology of human leishmaniasis in Greece, 1981-2011. Euro Surveill 18(29):pii = 20532
Gramiccia M (2011) Recent advances in leishmaniosis in pet animals: epidemiology, diagnostics and anti-vectorial prophylaxis. Vet Parasitol 181:23–30
Gray CD, Kinnear PR (2012) IBM SPSS statistics 19 made simple. Psychology Press
Hajjaran H, Mohebali M, Mamishi S, Vasigheh F, Oshaghi MA, Naddaf S R, Teimouri A, Edrissian G H, Zarei Z (2013) Molecular identification and polymorphism determination of cutaneous and visceral leishmaniasis agents isolated from human and animal hosts in Iran. BioMed Res Int: 789326
Hasegawa M, Kishino H, Yano T (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics Oxf Engl 17:754–755
Jamjoom MB, Ashford RW, Bates PA, Chance ML, Kemp SJ, Watts PC, Noyes HA (2004) Leishmania donovani is the only cause of visceral leishmaniasis in East Africa; previous descriptions of L. infantum and “L. archibaldi” from this region are a consequence of convergent evolution in the isoenzyme data. Parasitology 129:399–409
Jiménez M, González E, Iriso A, Marco E, Alegret A, Fúster F, Molina R (2013) Detection of Leishmania infantum and identification of blood meals in Phlebotomus perniciosus from a focus of human leishmaniasis in Madrid, Spain. Parasitol Res 112:2453–2459
Jiménez M, González E, Martín-Martín I, Hernández S, Molina R (2014) Could wild rabbits (Oryctolagus cuniculus) be reservoirs for Leishmania infantum in the focus of Madrid, Spain? Vet Parasitol 202:296–300
Kouam MK, Diakou A, Kanzoura V, Papadopoulos E, Gajadhar AA, Theodoropoulos G (2010) A seroepidemiological study of exposure to Toxoplasma, Leishmania, Echinococcus and Trichinella in equids in Greece and analysis of risk factors. Vet Parasitol 170:170–175
Kuhls K, Mauricio I, Pratlong F, Presber W, Schonian G (2005) Analysis of ribosomal DNA internal transcribed spacer sequences of the Leishmania donovani complex. Microbes Infect 7:1224–1234
Leite RS, Ferreira S de A, Ituassu LT, de Melo MN, de Andrade ASR (2010) PCR diagnosis of visceral leishmaniasis in asymptomatic dogs using conjunctival swab samples. Vet Parasitol 170: 201–206
Millán J, Ferroglio E, Solano-Gallego L (2014) Role of wildlife in the epidemiology of Leishmania infantum infection in Europe. Parasitol Res 113:2005–2014
Molina R, Jiménez MI, Cruz I, Iriso A, Martín-Martín I, Sevillano O, Melero S, Bernal J (2012) The hare (Lepus granatensis) as potential sylvatic reservoir of Leishmania infantum in Spain. Vet Parasitol 190:268–271
Moreno I, Álvarez J, García N, de la Fuente S, Martínez I, Marino E, Toraño A, Goyache J, Vilas F, Domínguez L, Domínguez M (2014) Detection of anti-Leishmania infantum antibodies in sylvatic lagomorphs from an epidemic area of Madrid using the indirect immunofluorescence antibody test. Vet Parasitol 199:264–267
Ntais P, Sifaki-Pistola D, Christodoulou V, Messaritakis I, Pratlong F, Poupalos G, Antoniou M (2013) Leishmaniases in Greece. Am J Trop Med Hyg 89:906–915
Parvizi P, Moradi G, Akbari G, Farahmand M, Ready PD, Piazak N, Assmar M, Amirkhani A (2008) PCR detection and sequencing of parasite ITS-rDNA gene from reservoirs host of zoonotic cutaneous leishmaniasis in central Iran. Parasitol Res 103:1273–1278
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics Oxf Engl 14:817–818
Ruiz-Fons F, Ferroglio E, Gortázar C (2013) Leishmania infantum in free-ranging hares, Spain, 2004-2010. Euro Surveill 18(30):pii = 20541
Schönian G, Akuffo H, Lewin S, Maasho K, Nylén S, Pratlong F, Eisenberger CL, Schnur LF, Presber W (2000) Genetic variability within the species Leishmania aethiopica does not correlate with clinical variations of cutaneous leishmaniasis. Mol Biochem Parasitol 106:239–248
Sobrino R, Ferroglio E, Oleaga A, Romano A, Millán J, Revilla M, Arnal MC, Trisciuoglio A, Gortázar C (2008) Characterization of widespread canine leishmaniasis among wild carnivores from Spain. Vet Parasitol 155:198–203
Souza TD, Turchetti AP, Fujiwara RT, Paixão TA, Santos RL (2014) Visceral leishmaniasis in zoo and wildlife. Vet Parasitol 200:233–241
Tabachnick BG, Fidell LS (2007) Using multivariate statistics. Allyn & Bacon/Pearson Education, Boston
Wang J-Y, Gao C-H, Yang Y-T, Chen H-T, Zhu X-H, Lv S, Chen S-B, Tong S-X, Steinmann P, Ziegelbauer K, Zhou X-N (2010) An outbreak of the desert sub-type of zoonotic visceral leishmaniasis in Jiashi, Xinjiang Uygur Autonomous Region, People’s Republic of China. Parasitol Int 59:331–337
Yang B-B, Guo X-G, Hu X-S, Zhang J-G, Liao L, Chen D-L, Chen J-P (2010) Species discrimination and phylogenetic inference of 17 Chinese Leishmania isolates based on internal transcribed spacer 1 (ITS1) sequences. Parasitol Res 107:1049–1065
Zemanová E, Jirků M, Mauricio IL, Miles MA, Lukes J (2004) Genetic polymorphism within the Leishmania donovani complex: correlation with geographic origin. Am J Trop Med Hyg 70:613–617
Acknowledgments
This research has been co-financed by the European Union (European Social Fund—ESF) and Greek national funds through the Operational Program "Education and Lifelong Learning" of the National Strategic Reference Framework (NSRF)-Research Funding Program: THALES, Investing in knowledge society through the European Social Fund. We would like to thank the personnel of the Hellenic Center for Diseases Control and Prevention (HCDCP) for providing the surveillance data on human Leishmaniasis cases: Danae Pervanidou, Eleni Triantafyllou, and Theano Georgakopoulou.
Conflict of interest
The authors declare that they have no competing interests.
Compliance with ethical standards
The hare samples examined for the presence of Leishmania DNA in this study represent archived material collected opportunistically (no active capture, killing, and sampling of wild animals specifically for this study was performed) from animals hunter-harvested by members of the Greek Hunting Federation of Macedonia and Thrace, during the hunting seasons, according to the prerequisites of the Greek Legislation (Ministerial Decision 103305/6093/14-11-2007, FEK 1626/Β΄/13.8.2008, FEK 1611/Β΄/2009, FEK 1183/6-8-2010, FEK 1763/4-8-2011), or from hares found dead and submitted to our laboratory for Passive Wildlife Disease surveillance during the period 2007–2011. Thus, special approval was not necessary, and steps to ameliorate suffering were not applicable in this study. Research on animals as defined in the EU Ethics for Researchers document (European Commission, 2007, Ethics for Researchers—Facilitating Research Excellence in FP7, Luxembourg: Office for Official Publications of the European Communities, ISBN 978-92-79-05474-7) is not a part of the study.
The canine samples included in this study were collected by private practicing veterinarians. No animals were euthanized during the study and efforts were taken to ameliorate animal suffering. The study did not involve any experimentation but was based on samples that had been collected from the dogs for routine diagnostic purposes. Diagnostic veterinary procedures are not within the context of relevant EU legislation for animal experimentations (Directive 86/609/EC) and may be performed in order to diagnose animal diseases and improve animal welfare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tsokana, C.N., Sokos, C., Giannakopoulos, A. et al. First evidence of Leishmania infection in European brown hare (Lepus europaeus) in Greece: GIS analysis and phylogenetic position within the Leishmania spp. Parasitol Res 115, 313–321 (2016). https://doi.org/10.1007/s00436-015-4749-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-015-4749-8