
Abstract
Ninety-two Cryptosporidium sp.-positive fecal samples of dairy diarrheic or non-diarrheic calves from 30 cattle herds in Normandy (France) were selected. Here, the aim was to investigate the species of Cryptosporidium excreted as well as the subtypes of Cryptosporidium parvum found in 7–17-day-old dairy calves. Excretion levels were comprised between 2 × 104 and 4 × 107 oocysts per gram of feces. Here, a nested 18S SSU rRNA PCR associated with sequencing was performed for identification of Cryptosporidium species and revealed the presence of C. parvum in most cases (80/82), except for two animals which were infected with Cryptosporidium bovis. Then, C. parvum samples were submitted to gp60 PCR. For 39 samples from 24 different herds, a multilocus analysis based on four mini-microsatellites loci (MM19, MM5, MSF, and MS9-Mallon) were conducted. These results were combined with sequence analysis of the gp60 to obtain multilocus types (MLTs). Here, C. parvum gp60 genotyping identified three subtypes in the IIa zoonotic allele family: IIaA15G2R1 (88 %), IIaA16G3R1 (10 %), and IIaA19G2R1 (2 %), and we identified 12 MLTs. The MS9-Mallon locus was reported as the most polymorphic (five alleles). The most common MLT was MLT 1 with 15 samples in 10 farms: (MS9-M: 298, MSF: 165, MM5: 264, MM19: 462, and gp60 subtype: IIaA15G2R1). When comparing diarrheic and non-diarrheic fecal samples, no difference was seen for distribution of Cryptosporidium species, C. parvum gp60 subtypes, and MLTs. Here, in a range of oocyst excretion of 104–107 opg, both in diarrheic and non-diarrheic calves, infection was mainly due to C. parvum and to the zoonotic subtype: IIaA15G2R1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cryptosporidium is a protozoan parasite of medical and veterinary importance recognized as an important zoonotic pathogen that infects a wide host range (mammals, birds, reptiles, amphibians, and fish), including humans (Fayer 2010; Xiao 2010). This parasite is the agent of cryptosporidiosis, a disease characterized by diarrhea in human and livestock. Currently, no drug therapy is yet available and the high resistance of oocysts of Cryptosporidium spp. in the environment makes cryptosporidiosis difficult to control (Caccío and Pozio 2006).
The first reported case of cryptosporidiose in cattle was in 1971 (Panciera et al. 1971); now, bovine cryptosporidiosis is of great concern due to the huge number of cattle and their economic importance. In fact, Cryptosporidium spp. is one of the major causes of neonatal calf diarrhea, while in yearling heifers and mature cows, asymptomatic infection commonly occurs (Santín et al. 2008). Economic losses due to cryptosporidiosis in farm animals are related to morbidity and mortality according to Cryptosporidium species/genotypes causing the infection. Thanks to molecular characterization, several studies demonstrated that cattle could be infected with different species: Cryptosporidium parvum, Cryptosporidium bovis, Cryptosporidium ryanae, and Cryptosporidium andersoni. The three former species were found in most age categories (Kváč et al. 2006; Fayer et al. 2008). However, C. parvum was predominantly found in pre-weaned dairy calves (<2 months of age) while C. bovis and C. ryanae were mainly observed in post-weaned dairy calves (>2 months of age) (Santín et al. 2004; Fayer et al. 2005). C. andersoni was dominantly found in adult cows (Feng et al. 2007; Santín et al. 2008; Fayer 2010; Xiao 2010). More recent studies identified the species C. bovis and C. ryanae in younger dairy or beef calves (<2 month of age) (Silverlås et al. 2010; Silverlås and Penedo-Blanco 2012; Rieux et al. 2013). As far as pathogenic aspects are concerned, it has been shown that C. parvum infecting neonates is responsible for diarrhea, dehydration, and growth retardation in calves whereas animals infected with other Cryptosporidium species usually exhibit no overt clinical signs (Kváč and Vítovec 2003; Fayer et al. 2008).
Over the last few years, typing and sub-typing techniques have been applied to understand the dynamics of C. parvum transmission between cattle and human populations (Alves et al. 2006; Plutzer and Karanis 2009). At least 11 different C. parvum families with many subtypes have been described on the basis of sequence analysis of a 60-kDa glycoprotein (gp60) (Xiao 2010). Moreover, previous studies have shown that cattle could be considered as a primary reservoir for Cryptosporidium oocysts for zoonotic subtypes of C. parvum belonging to families IIa and IId, so infected calves could be a risk factor for the public health via environmental contamination from their manure being spread on farmland or their grazing on watersheds (Monis and Thompson 2003; Xiao and Fayer 2008; Chalmers and Giles 2010).
Sequencing of the 60-kDa glycoprotein is currently the most widely used sub-typing method, due to the gene heterogeneity, but other methods based on the characterization of short variable number tandem repeats, known as microsatellites and minisatellites, are being increasingly used in multilocus analysis to investigate the genetic structure and or geographic tracking of Cryptosporidium (Jex et al. 2008; Xiao 2010). Methods used to investigate polymorphisms of micro-minisatellites are based on fragment size analysis of PCR-amplified products by sequencing (Chalmers et al. 2005; Wielinga et al. 2008), capillary electrophoresis (CE) with labeled primers (Mallon et al. 2003; Morrison et al. 2008), and polyacrylamide or precast high-resolution slab gel (Feng et al. 2000; Tanriverdi and Widmer 2006; Tanriverdi et al. 2006). These different approaches to identify alleles were compared by Díaz et al. (2012); these authors showed that CE approach was an economic and rapid option for performing fragment size analysis. Numerous loci have been investigated in multilocus genotyping study references but until now no markers have been universally adopted (Robinson and Chalmers 2012).
Available data concerning molecular epidemiology of Cryptosporidium in calves, in France, are quite limited: only two longitudinal studies were conducted, one in dairy calves in Brittany (Follet et al. 2011) and one in beef calves in Deux-Sèvres (Rieux et al. 2013).
Here, we wanted to investigate which species of Cryptosporidium were excreted by young diarrheic and non-diarrheic calves (7–17 days of age) from a wide range of farms located in a specialized dairy area in western France. Then, we further characterized the C. parvum isolates through genotyping and sub-typing with a multilocus analysis using CE analysis combined to a result of a sequence of the gp60 gene to observe the variability of multilocus types (MLTs) among the location of farms and to observe the possibility of one genotype or MLT to be associated to clinical signs.
Materials and methods
Fecal sample collection
An epidemiological study was conducted in 100 dairy herds in a dairy specialized department in Normandy (France): 10 calves <21 days of age were sampled once/herd. Feces were collected directly for the rectum using plastic gloves. For each animal, the number of animal identification, the age, the sampling date, and the clinical status were recorded. These samples were transported to the laboratory in a sample pot and then stored at 4 °C for a maximum of 48 h before analysis. Each sample was screened for Cryptosporidium spp. infection using a semiquantitative fecal smear staining (Heine 1982). Samples were done in compliance with the animal welfare and did not cause any pain according to the ethics committee for animal experimentation no. 16 (French Referential)
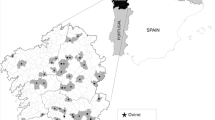
For the present study, a selection of farms was made to obtain contrasted both epidemiological and clinical situations. From the 100 farms, we selected 30 farms, within a 140-km radius (Fig. 1) as following: we chose 15 dairy cattle herds with four or more diarrheic calves (4 out of 10 was the median number of diarrheic calves farm in the whole survey), representing the herds with a high prevalence of diarrhea and 15 cattle herds with less than 4/10 diarrheic calves, representing the herds with low prevalence of diarrhea. We selected 50 diarrheic calves positive for Cryptosporidium aged 7 to 17 days from 15 farms and we selected 42 non-diarrheic, but positive for Cryptosporidium, calves aged 8 to 17 days from 15 farms (Fig. 1). The fecal scores of the selected samples for Cryptosporidium oocyst were comprised between four and five corresponding to a high level of excretion by Heine technique.

Map of the department of Orne (north-western France) showing the location of farms included in this study. The farm with diarrheic animals were represented by a dark stars and the farms with non-diarrheic animals were represented with a gray stars
Sample processing (oocyst concentration and immunofluorescence test)
All samples, except 16 frozen samples which were directly submitted to DNA extraction, were examined by immunofluorescence test (IFT) after oocyst concentration. One gram of feces was used for the oocyst concentration using ethyl acetate as previously described (Castro-Hermida et al. 2005). Aliquots of 10 μl of the sediment were fixed on slides using acetone at 4 °C for 10 min and processed using an IFT commercial kit (Merifluor® Cryptosporidium/Giardia, Meridian Bioscience Europe, Nice, France). The samples were observed by fluorescence microscopy using ×400 magnification. The number of oocysts per gram of feces was given by [number of oocysts seen on slide / (volume of sample examined (ml) × weight of feces (g))]. The limit of detection of this technique was estimated at 100 opg (Fayer et al. 2000). Positive samples were then submitted to molecular characterization.
DNA isolation
Genomic DNA was extracted from 500 μl of oocyst suspension using an automatic extractor (Maxwell® MDX 16, Promega) with a tissue kit (Maxwell® 16 Tissue DNA Purification Kit AS1030), after a preliminary step of breaking achieved with a RiboLyser™ (Bio-Rad®).
PCRs amplification of 18S SSU rRNA gene and gp60 gene for the species identification and sub- genotyping of C. parvum
All positive samples by IF were submitted to species identification and sub-typing using gp60. A nested PCR protocol was used to amplify an 830-bp fragment of the 18S rRNA gene. The nested polymerase chain reaction protocol was performed in two steps according to Xiao et al. (1999, 2001). The specific used primers were F1forward 5′-TTCTAGAGCTAATA CATGCG-3′and R1 reverse 5′-CCCATTTCCTTCGAAACAGGA-3′ for primary PCR and F2 forward 5′-GGAAGGGT TGTATTTATTAGATAAAG-3′ and R2 reverse 5′-AAGGA GTAAGGAACAACCTCCA3′ for secondary PCR.
For samples identified as C. parvum, a nested PCR protocol was used to amplify a 850-bp fragment of the gp60 to identify the subtype. The nested polymerase chain reaction protocol was performed in two steps according to Gatei et al. (2007). The specific used primers were gp60F forward 5′-ATAGTCTCCGCTGTATTC-3′ and gp60R1 reverse 5′-GGAAGGAAC GATGTATCT-3′ for primary PCR and gp60F2 forward 5′-TCCGCTGTATTCTCAGCC-3′ and gp60R2 reverse 5′-GCAGAGGAACCAGCATC-3′ for secondary PCR. These amplifications were performed on an iCycler Thermal Cycler from Bio-Rad®. Amplification products (10 μL) were separated on 2 % agarose and stained with ethidium bromide. PCR was repeated at a maximum of two times for samples that were positive by IF but remained negative by PCR.
Sequence analysis
All the secondary PCR products obtained, after amplification of the 18S rRNA gene or gp60 gene, were sequenced in both directions. DNA sequencing reactions were performed using internal primers of the nested PCR using an ABI 3730XL sequencer (Applied Biosystems, Warrington, UK) by Genoscreen (Lille, France). The sequence alignment was checked for sequencing accuracy using the software BioEdit Sequence Alignment Editor (version 7.0.9.0). The sequences obtained of each strand were aligned and then were compared with published sequences in the GenBank database using Basic Local Alignment Search Tool [NCBI (http://www.ncbi.nlm.nih.gov/BLAST)].
Multilocus analysis
All samples which had a gp60 sequence of C. parvum available (20 samples from diarrheic calves and 19 samples from non-diarrheic calves) were submitted to multilocus analysis. These 39 samples were collected in 24 farms located within a 140-km radius.
Molecular characterization
The four polymorphic loci used in this study were as follows: MS9-Mallon, MM19, MSF, and MM5. These markers were chosen from the literature to observe if there were some differences of MLT according to the location of farms or clinical signs observed in these animals (Mallon et al. 2003; Morrison et al. 2008; Robinson and Chalmers 2012). These markers were the more informative markers concerning the multilocus analysis of C. parvum isolates (Robinson and Chalmers 2012). The MS9-Mallon marker contains a TGGACT repeat in a 2,016-bp gene encoding a hypothetical protein located at position 640137 to 642152 on chromosome 5. The MM19 marker contains a GGA (G/T)C(T/A) repeat in a 7,230-bp gene located at position 1208520 to 1215749 on chromosome 8. The MSF marker contains a TCCTTCCTGAGC repeat in gene encoding a signal peptide and the MM5 marker contains a TCCTCCTCT repeat located in a 11,418-bp gene located at positions 1002285 to 1013702 on chromosome 6. The sequences of the primers used for amplification were described by Mallon et al. (2003) and Morrison et al. (2008). The sequences of the primers used were the following:
-
MS9-Mallon-F, 5′-GGACTAGAAATAGAGCTTTGGCTGG-3′;
-
MS9-Mallon-R, 5′- GTCTGAGACAGAATCTAGG ATCTAC-3′;
-
MSF-F, 5′- TCGGCCTCCTCTACAG-3′;
-
MSF-R, 5′- AGAAGAAAGCCAA GAAGGGT-3′;
-
MM19-F, 5′- GATT CTGTCAACTTTGAATTCAG-3′;
-
MM19-R, 5′ CCAACC CCGAATTCATTTCCAAC-3′;
-
MM5-F, 5′ GGAGAAGATAAGCTAGCCGAATCT-3′;
-
MM5-R, 5′ CCTGGACTTGGATTTGGACTTACACC-3′.
All forwards primers were labeled with a fluorophore and all reactions were undertaken by Genoscreen (Lille, France). Ten characterized samples providing for another study, acting for positive control, were sent for testing the primers of the different markers and the CE technique before the amplification test for the 39 samples.
Allele and multilocus type identification
The size of each PCR product was estimated by electrophoresis on a capillary apparatus by Genoscreen (Lille, France) by comparison to size standards which allowed us in determining size of alleles by software. The software automatically assigned fragment sizes to the resulting electropherogram peaks; allele size was deduced from that of the main peak rounded to the nearest probable base pair number. Each allele was assigned a number and an MLT designated by the combination of alleles at each locus. If one isolate contained more than one allele at one locus, this was the result of the presence of mixed infection.
Results
Selection of samples and quantification of the level of excretion
Sampling characteristics and oocyst excretion range were displayed on Tables 1 and 2. From each farm, we selected between 1 and 6 diarrheic calves (herds with high prevalence of diarrhea) and between 1 and 5 non-diarrheic calves (herds with low prevalence of diarrhea). Animals were sampled once before 21 days of life. Calves chosen for the present study have an average age of 11 days for the diarrheic calves and an average age of 13 days for the non-diarrheic ones. The level of excretion of these samples was accurately quantified by IFT and comprised between 2 × 104 and 4 × 107 opg. For the 50 diarrheic samples, the average level of excretion was 2.9 × 106 opg (varying between 3 × 104 and 3 × 107 opg), and for the 42 non-diarrheic samples, the average level was 3 × 106 opg (varying between 2 × 104 and 4 × 107 opg).
Cryptosporidium species identification by 18S rRNA sequence analysis
Eighty-two isolates (45 diarrheic and 37 non-diarrheic) were amplified by PCR 18S (Tables 1 and 2). All sequences from diarrheic calves were exploited and all isolates were identified as C. parvum (Table 1). Among the 37 non-diarrheic isolates, 35 were identified as C. parvum and 2 were identified as C. bovis (Table 2). Sequence analysis revealed between 99 and 100 % identity with the sequences JX416362.1 and JX559849.1 for isolates of C. parvum and between 99 and 100 % identity with the sequence JX515546.1 of GenBank for the two isolates of C. bovis.
C. parvum sub-typing by gp60 sequence analysis
From the 80 samples identified as C. parvum, 52 samples were successfully amplified by gp60 PCR. DNA sequences of isolates from diarrheic calves (31/45) and non-diarrheic animals (21/35) were analyzed. Sequence analysis of the gp60 gene revealed between 99 and 100 % identity with the sequences JF727755.1, HQ005741.1, and JQ362495.1 of GenBank corresponding to the subtypes IIaA15G2R1 (46 isolates/52), IIaA16G3R1 (5/52), and IIaA19G2R1(1/52), respectively. The subtype IIaA15G2R1 was found in 21 farms, the subtype IIaA16G3R1 was found in two farms, and the subtype IIaA19G2R1 was found in one single farm. Interestingly, only one subtype was identified in each farm (Tables 1 and 2).
Multilocus typing
In a first step, 10 positive controls (samples containing between 105 and 107 opg, from diarrheic dairy calves identified C. parvum (subtype IIaA15G2R1) used in another study) were sent to Genoscreen for testing the primers of the different markers and the CE technique. Then, we sent 39 samples (20 diarrheic and 19 non-diarrheic) from 24 dairy cattle herds. The level of excretion of these 39 samples comprised between 3 × 104 and 4 × 107 opg, and among these isolates, 37 were identified with the subtype IIaA15G2R1 from 20 farms and 2 with the subtype IIaA16G3R1from 2 farms.
A total of 38 samples were typed at MM19 and MM5, four alleles were found for MM19 and four for MM5. A total of 37 samples were typed at MS9-Mallon and MSF with five alleles for MS9-Mallon and two for MSF (Table 3). MS9-Mallon was the most polymorphic marker.
A total of 36 C. parvum isolates were typable at all four loci and used for multilocus analysis. Twelve MLTs were identified with the 31 gp60 subtype IIaA15G2R1 isolates being differentiated into 10 MLTs and the 2 gp60 subtype IIaA16G3R1 isolates being differentiated into 2 MLTs (Tables 3 and 4). Moreover, in two farms (<4 diarrheic calves, herd numbers 4 and 15), two different profiles of MLT were observed. In addition, three samples (10, 8 %) contained more than one allele at one locus resulting in the presence of mixed infection.
The MLT1 was the most prevalent type in our study (15 calves in 10 farms) and this profile was equally found in diarrheic and non-diarrheic samples. Here, no difference regarding MLT found was seen between diarrheic and non-diarrheic samples.
Discussion
Bovine cryptosporidiosis is one of the major causes of neonatal calf diarrhea and young calves (<1 month of age) are frequently infected with Cryptosporidium sp. (Quílez et al. 1996). In France, only two recent studies have included molecular tools for the study of Cryptosporidium prevalence in calves (Follet et al. 2011; Rieux et al. 2013). The present study is the first one in France to extend molecular characterization (18S and gp60 combined to MLT analysis) to describe genetic characteristics of Cryptosporidium in 92, 7–17 days old dairy calves coming from 30 cattle herds.
Our results showed that C. parvum was the dominant species identified (80/82) and confirmed the previous data of Follet et al. in Brittany in 2011 where C. parvum caused 86.7 % of Cryptosporidium infection in 5-week-old calves. Other studies conducted worldwide, in dairy calves, also showed that C. parvum was responsible for more than 80 % of Cryptosporidium infection (Plutzer and Karanis 2009; Santín et al. 2004; Brook et al. 2009).
The two other isolates collected from non-diarrheic calves were identified as C. bovis. This species is mainly found in weaned animals (Santín et al. 2004, 2008; Coklin et al. 2009; Fayer 2010; Xiao 2010). C. bovis has been recently recorded in beef and dairy calves in France and Sweden, from 7 or 9 days of age with high level of excretion (up to 8 × 106 opg) (Silverlås et al. 2010; Rieux et al. 2013).
Differences in pathogenicity between the subfamilies of Cryptosporidium hominis have been described by Cama et al. (2007) with some subfamily as Id that was associated with diarrhea whereas others as subfamilies Ia, Ib, or Ie that was not. In our study, we searched for such differences according to the location of dairy cattle herds or to the presence or absence of clinical signs like it. The results concerning the sequence analysis of the gp60 gene showed three subtypes belonging to the zoonotic allele family IIa: IIaA15G2R1 (88 %), IIaA16G3R1 (10 %), and IIaA19G2R1 (2 %) subtypes were detected. Subtypes IIaA15G2R1 and IIaA16G3R1 were found in diarrheic and non-diarrheic animals like it was already described in literature (Brook et al. 2009; Xiao 2010; Follet et al. 2011). Follet et al. (2011) found a higher subtype diversity compared to our study with six different subtypes of C. parvum (IIaA15G2R1, IIaA17G1R1, IIaA16G3R1, IIaA16G2R1, IIaA16G1R1, and IIaA13G1R1) in 15 cattle herds (fattening units) in Brittany by analyzing the gp60 gene. The present study was conducted in one single area (department), in 30 cattle herds located within a 140-km radius, without calf movements between herds, what is quite different from the situation of Follet et al. (2011), where calves coming from a wider geographic area (six departments) were collected in 15 fattening units possibly explaining the difference in number of subtypes found. Moreover, the number of samples in the study of Follet et al. (2011) was higher. Our results are in agreement with several previous studies that described IIaA15G2R1 as the most widely IIa subtype distributed in calves worldwide (Xiao 2010). Interestingly, in some countries (Slovenia, Portugal, and The Netherlands), it has been shown that this subtype was predominant in human cryptosporidiosis outbreaks (Alves et al. 2006; Wielinga et al. 2008; Soba and Logar 2008). Such data are not available in France. The second subtype found in this work, IIaA16G3R1, has also been described with a similar frequency in cattle in The Netherlands Wielinga et al. (2008), England (Brook et al. 2009), and in France (Follet et al. 2011). The last subtype found in this study, IIaA19G2R1, has been recorded with high figures (7 %) in diarrheic calves less than 2 months of age in 20 farms in Australia (Ng et al. 2012). In previous studies about subtype characterization as well as in the present work, a single subtype was found per herd (Alves et al. 2006; Santín et al. 2008). This suggests a stable within herd clonal pattern in the C. parvum population (Soba and Logar 2008). In contrast, in area with animal exchange between herds, more than one subtype can be found per farm unit (Brook et al. 2009; Follet et al. 2011; Silverlås et al. 2013).
The majority of the multilocus analyses targeting microsatellite and/or minisatellite regions have been used to investigate the population structure, to understand the transmission dynamics of some strains of Cryptosporidium spp. and figure out the host–parasite interaction (Mallon et al. 2003; Tanriverdi and Widmer 2006; Tanriverdi et al. 2006; Morrison et al. 2008; Robinson and Chalmers 2012). Here, four markers considered as informative for the species C. parvum were added to results of gp60 typing. Results of the present study reveal that the marker MS9-Mallon was the most polymorphic and highly contributed to the discriminatory power of the multilocus approach. The combination of polymorphisms at the four loci generated a total of 12 MLTs among the C. parvum isolates from dairy pre-weaned calves, diarrheic and non-diarrheic. In Italy, a recent largest MLT study (northern, central, and southern Italy) used seven polymorphic markers, whose MM19, MS9-Mallon, and MM5, and was conducted with numerous and diverse samples (122 isolates of calves, 21 of lambs, 21 of goats, and 14 of humans) (Drumo et al. 2012). The authors found 102 MLTs and found that MM19 marker was the most polymorphic closely followed by the MS9-Mallon marker. Another multilocus study conducted, in Spain, with three other markers identified 9 MLT from 41 C. parvum isolates (Díaz et al. 2012). In this last study, samples of calves and lambs were tested to search for differences between subtypes according to the host. The authors concluded that one MLT was predominant in cattle and another MLT was predominant in lambs. In our study, MLT was done within a host (calves) and in a more restricted geographical area including several distinct herds facing clinical cryptosporidiosis or not. Concerning the numbers of markers used and the MLTs found, our results are in general agreement with these previous studies. Here, the predominant MLT was MLT1 and it was found in 15 calves from 10 farms (nine diarrheic calves belonging to five herds and in six non-diarrheic calves belonging to five other herds). We also found 10.8 % of C. parvum isolates contained more than one allele at one locus resulting in the presence of mixed infection. Here, we also observe two different MLTs in one cattle. The level of mixed infection in C. parvum populations varies in published data from no detection in New Zealand (19 humans and 7 calf isolates) to 8.2 % to 30 % in Serbia (23 calves) and to 11.6 % in Italy (Tanriverdi and Widmer 2006; Tanriverdi et al. 2006; Morrison et al. 2008; Drumo et al. 2012). Concerning MLT analysis, numerous loci have been investigated but until now no marker sets have been universally adopted, producing a fragmented view of the genetic variability, hindering data comparison and making meta-analyses impossible (Robinson and Chalmers 2012). Additionally, different approaches to identifying alleles have been used (DNA sequencing, CE, and high-resolution slab gel electrophoresis) and there is no consensus in the methods so far.
Here, no differences were seen concerning the species of Cryptosporidium identified in diarrheic or non-diarrheic samples as C. parvum was mainly found. This is in agreement with previous literature for young dairy calves (Santín et al. 2004; Plutzer and Karanis 2009; Trotz-Williams et al. 2007; Santín et al. 2008). Others pathogens could be found in diarrheic samples such as bacteria or virus in association or not with Cryptosporidium but this aspect has not been included in our study. It is also important to mention that we worked only with samples highly loaded in oocysts of Cryptosporidium and that dairy calves were sampled once. It is therefore possible that some animals had clinical signs before or after the date of collection. In fact, selected animals have an average age of 11 days for the diarrheic animals and average age of 13 days for the non-diarrheic animals. It has been shown that the prevalence of excretion of Cryptosporidium oocysts was highest in pre-weaned animals with excretion beginning at 4–5 days of age, reaching a peak between 7 and 10 days and declining beyond 16 and 17 days (Trotz-Williams et al. 2007). In addition, we did not observe differences in subtypes of C. parvum (IIaA15G2R1) or in the MLT’s (MLT1predominant with MS9-M: 298, MSF: 165, MM5: 264, MM19: 462, and gp60 subtype: IIaA15G2R1) according to the diarrheic or non-diarrheic status of calves.
Finally, this study shows that young dairy calves (7–17 days of age) are mainly infected by the species: C. parvum and by the zoonotic subtype: IIaA15G2R1. Moreover, we observed that whatever the level of characterization chosen, species identification, sub-typing, or multilocus analyses, no difference was seen in Cryptosporidium isolates according to the location of farms or to the clinical status.
References
Alves M, Xiao L, Antunes F, Matos O (2006) Distribution of Cryptosporidium subtypes in humans and domestic and wild ruminants in Portugal. Parasitol Res 99:287–292
Brook EJ, Anthony Hart C, French NP, Christley RM (2009) Molecular epidemiology of Cryptosporidium subtypes in cattle in England. Vet Journal 179:378–382
Caccío SM, Pozio E (2006) Advances in the epidemiology, diagnosis and treatment of cryptosporidiosis. Expert Rev Anti Infect Ther 4:429–443
Cama VA, Ross JM, Crawford S, Kawai V, Chavez-Valdez R, Vargas D, Vivar A, Ticona E, Navincopa M, Williamson J, Ortega Y, Gilman RH, Bern C, Xiao L (2007) Differences in clinical manifestations among Cryptosporidium species and subtypes in HIV-infected persons. J Infect Dis 196:684–691
Castro-Hermida JA, Pors I, Poupin B, Ares-Mazás E, Chartier C (2005) Prevalence of Giardia duodenalis and Cryptosporidium parvum infections in goat kids in western France. Small Rumin Res 56:259–264
Chalmers RM, Giles M (2010) Zoonotic cryptosporidiosis in the UK—challenge for control. J Appl Microbiol 109:1487–1497
Chalmers RM, Ferguson C, Cacciò S, Gasser RB, Abs El-Osta YG, Heijnen L, Xiao L, Elwin K, Hadfield S, Sinclair M, Stevens M (2005) Direct comparison of selected methods for genetic categorisation of Cryptosporidium parvum and Cryptosporidium hominis species. Int J Parasitol 35:397–410
Coklin T, Uehlinger FD, Farber JM, Barkema HW, O'Handley RM, Dixon BR (2009) Prevalence and molecular characterization of Cryptosporidium spp. in dairy calves from 11 farms in Prince Edward Island. Canada Vet Parasitol 160:323–326
Díaz P, Hadfield SJ, Quílez J, Soilán M, López C, Panadero R, Díez-Baňos P, Morrondo P, Chalmers RM (2012) Assessment of three methods for multilocus fragment typing of Cryptosporidium parvum from domestic ruminants in north west Spain. Vet Parasitol 186:188–195
Drumo S, Widmer G, Morrison LJ, Tait A, Grelloni V, D’Avino N, Pozio E (2012) Evidence of host-associated populations of Cryptosporidium parvum in Italy. Appl Enviro Microbiol 78:3523–3529
Fayer R (2010) Taxonomy and species delimitation in Cryptosporidium. Exp Parasitol 124:90–97
Fayer R, Trout JM, Graczyk TK, Lewis EJ (2000) Prevalence of Cryptosporidium, Giardia and Eimeria infections in post-weaned and adult cattle on three Maryland farms. Vet Parasitol 93:103–112
Fayer R, Santín M, Xiao L (2005) Cryptosporidium bovis n. sp. (Apicomplexa: Cryptosporidiidae) in cattle (Bos taurus). J Parasitol 91:624–629
Fayer R, Santin M, Trout JM (2008) Cryptosporidium ryanae n.sp. (Apicomplexa: Cryptosporidiidae) in cattle (Bos taurus). Vet Parasitol 156:191–198
Feng X, Rich SM, Akiyoshi D, Tumwine JK, Kekitiinwa A, Nabukeera N, Tzipori S, Widmer G (2000) Extensive polymorphism in Cryptosporidium parvum identified by multilocus microsatellite analysis. Appl Environ Microbiol 66:3344–3349
Feng Y, Ortega Y, He G, Das P, Xu M, Zhang X, Fayer R, Gatei W, Cama V, Xiao L (2007) Wide geographic distribution of Cryptosporidium parvum and deer like genotype in bovines. Vet Parasitol 144:1–9
Follet J, Guyot K, Leruste H, Follet-Dumoulin A, Hammouma-Ghelboun O, Certad G, Dei Cas E, Halama P (2011) Cryptosporidium infection in a veal calf cohort in France: molecular characterization of species in longitudinal study. Vet Res 42:116
Gatei W, Das P, Dutta P, Sen A, Cama V, Lal AA, Xiao L (2007) Multilocus sequence typing and genetic structure of Cryptosporidium hominis from children in Kolkata, India. Infect Genet Evol 2:197–205
Heine J (1982) Eine einfache Nachweismethod fur Kryptosporidien in Kot. Zentralbl Veterinaermed Reihe B 29:324–327
Jex AR, Smith HV, Monis PT, Campbell BE, Gasser RB (2008) Cryptosporidium-biotechnical advances in the detection, diagnosis and analysis of genetic variation. Biotechnol Adv 26:304–317
Kváč M, Vítovec J (2003) Prevalence and pathogenicity of Cryptosporidium andersoni in one herd of beef cattle. J Vet Med B infect Dis Vet Public Health 50:451–457
Kváč M, Kouba M, Vítovec J (2006) Age-related and housing-dependence of Cryptosporidium infection of calves from dairy and beef herds in South Bohemia, Czech Republic. Vet Parasitol 137:202–209
Mallon M, MacLeod A, Wasling J, Smith H, Reilly B, Tait A (2003) Population structures and the role of genetic exchange in the zoonotic pathogen Cryptosporidium parvum. J Mol Evol 56:407–417
Monis PT, Thompson RC (2003) Cryptosporidium and Giardia-zoonoses: fact or fiction? Infect Genet Evol 3:233–244
Morrison JL, Mallon EM, Smith VH, MacLeod A, Xiao L, Tait A (2008) The population structure of the Cryptosporidium parvum population in Scotland: a complex picture. Infect Genet Evol 8:121–129
Ng J, Eastwood K, Walker B, Durrheim DN, Massey PD, Porigneaux P, Kemp R, McKinnon B, Laurie K, Miller D, Bramley E, Ryan U (2012) Evidence of Cryptosporidium transmission between cattle and humans in northern New South Wales. Exp Parasitol 130:437–441
Panciera RJ, Thomassen RW, Garner FM (1971) Cryptosporidial infection in a calf. Vet Pathol 8:479–484
Plutzer J, Karanis P (2009) Genetic polymorphism in Cryptosporidium species: an update. Vet Parasitol 165:187–199
Quílez J, Sanchez-Acedo C, del Cacho E, Clavel A, Causape AC (1996) Prevalence of Cryptosporidium and Giardia infection in cattle in Aragon (northeastern Spain). Vet Parasitol 66:139–146
Rieux A, Chartier C, Pors I, Paraud C (2013) Dynamics of excretion and molecular characterization of Cryptosporidium isolates in pre-weaned French beef calves. Vet Parasitol 195(1–2):169–172
Robinson G, Chalmers RM (2012) Assessment of polymorphic genetic markers for multi-locus typing of Cryptosporidium parvum and Cryptosporidium hominis. Exp Parasitol 6475:1–16
Santín M, Trout J, Xiao L, Zhou L, Greiner E, Fayer R (2004) Prevalence and age related variation of Cryptosporidium species and genotypes in dairy calves. Vet Parasitol 122:103–117
Santín M, Trout JM, Fayer R (2008) A longitudinal study of cryptosporidiosis in dairy cattle from birth to 2 years of age. Vet Parasitol 155:15–23
Silverlås C, Penedo-Blanco I (2012) Cryptosporidium spp. in calves and cows from organic and conventional dairy herds. Epidemiol Infect 8:1–11
Silverlås C, De Verdier K, Emanuelson U, Mattsson JG, Björkman C (2010) Cryptosporidium infection in herds with and without calf diarrhoeal problems. Parasitol Res 107:1435–1444
Silverlås C, Bosaeus-Reineck H, Naslund K, Björkman C (2013) Is there a need for improved Cryptosporidium diagnostics in Swedish calves? Int J Parasitol 43:155–161
Soba B, Logar J (2008) Genetic classification of Cryptosporidium isolates from humans and calves in Slovenia. Parasitol 135:1236–1270
Tanriverdi S, Widmer G (2006) Differential evolution of repetitive sequences in Cryptosporidium parvum and Cryptosporidium hominis. Infect Genet Evol 6:113–122
Tanriverdi S, Markovics A, Arslan MO, Itik A, Shkap V, Widmer G (2006) Emergence of distinct genotypes of Cryptosporidium parvum in structured host populations. Appl Environ Microbiol 72:2507–2513
Trotz-Williams LA, Wayne Martin S, Leslie KE, Duffield T, Nydam DV, Peregrine AS (2007) Calf-level risk factors for neonatal diarrhea and shedding of Cryptosporidium parvum in Ontario dairy calves. Prev Vet Med 82:12–28
Wielinga PR, de Vries A, van der Goot TH, Mank T, Mars MH, Kortbeek LM, van der Giessen JW (2008) Molecular epidemiology of Cryptosporidium in human and cattle in the Netherlands. Int J Parasitol 38:809–817
Xiao L (2010) Molecular epidemiology of cryptosporidiosis: an update. Exp Parasitol 124:80–89
Xiao L, Fayer R (2008) Molecular characterization of species and genotypes of Cryptosporidium and Giardia and assessment of zoonotic transmission. Int J Parasitol 38:1239–1255
Xiao L, Morgan UM, Limor J, Escalante A, Arrowood M, Shulaw W, Thomson RC, Fayer R, Lal AA (1999) Genetic diversity within Cryptosporidium parvum and related Cryptosporidium species. Appl Environ Microbiol 65:3386–3391
Xiao L, Bern C, Limor J, Sulaiman I, Roberts J, Checkley W, Cabrera L, Gilman RH, Lal AA (2001) Identification of 5 types of Cryptosporidium parasites in children in Lima, Peru. J Infect Dis 183:492–497
Acknowledgments
A. Rieux is a grateful recipient of a grant from Anses/Region Poitou-Charentes. The authors acknowledge the cooperation of Marie-Christine Dupuy of GDS61, in the department of Orne, France, for the collection of fecal samples.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rieux, A., Chartier, C., Pors, I. et al. Molecular characterization of Cryptosporidium isolates from high-excreting young dairy calves in dairy cattle herds in Western France. Parasitol Res 112, 3423–3431 (2013). https://doi.org/10.1007/s00436-013-3520-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-013-3520-2