
Abstract
Although it has been known for many years that Trichinella spiralis initiates infection by penetrating the columnar epithelium of the small intestine, the mechanisms by which T. spiralis infective larvae recognize and invade the intestinal epithelial cells (IECs) are unknown. It is speculated that the molecular interactions between the parasite and host enterocytes may mediate the recognition and invasion of IECs by T. spiralis. However, no Trichinella proteins that interact with the enterocytes have been identified previously. The aim of this study was to identify Trichinella proteins that bind to IECs by screening a T7 phage display cDNA library constructed using messenger RNA from T. spiralis intestinal infective larvae. Following five rounds of biopanning, sequencing, and bioinformatics analysis, ten T. spiralis proteins (Tsp1–Tsp10) with significant binding to normal mouse IECs were identified. The results of the protein classification showed that six proteins (Tsp1, calcium-transporting ATPase 2 protein; Tsp4, ovochymase-1; Tsp6, T-complex protein 1 subunit eta; Tsp7, glycosyl hydrolase family 47; Tsp8, DNA replication licensing factor MCM3; and Tsp10, nudix hydrolase) of these T. spiralis proteins were annotated with putative molecular functions. Out of the six proteins, five have catalytic activity, four have binding activity, and one has transporter activity. Anti-Tsp10 antibodies prevented the in vitro partial invasion of IECs by infective larvae and the mice immunized with the recombinant phage T7-Tsp10 showed a 62.8 % reduction in adult worms following challenge with T. spiralis muscle larvae. Although their biological functions are not yet fully known, these proteins might be related to the larval invasion of host enterocytes. Future experiments will be necessary to ascertain whether these proteins play important roles in the recognition and invasion of host enterocytes. The construction and biopanning of Trichinella phage display libraries provide a novel approach for searching for candidate genes that are related to invasion and for identifying protein interactions between parasite and host.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Trichinellosis is a parasitic zoonosis caused by eating raw or undercooked meat contaminated with infective larvae of the nematode genus Trichinella. Human trichinellosis is an emerging/reemerging disease and has been reported in 55 countries around the world (Cui et al. 2006; Pozio 2007; Wang et al. 2012a, b). Infection occurs by the consumption of animal meat containing encapsulated Trichinella larvae. Once ingested, the muscle larvae (ML) are released from their capsules in the duodenum as the result of the action of the host’s digestive enzymes and are activated by exposure to the intestinal contents or bile. Then, the activated intestinal infective larvae penetrate into the epithelial cells of the host’s small intestine. Shortly thereafter, the larvae molt four times (10–28 h post infection, hpi) and mature into adults that mate and reproduce, yielding the next generation of larvae (Kang et al. 2012). The life cycle of Trichinella spiralis is completed when newborn larvae develop into ML and induce the transformation of muscle cells into nurse cells (Campbell 1983). It is well known that the invasion of the host intestinal epithelium by infective larvae is the first step in T. spiralis infection. Previous studies have shown that infective larvae invade the intestinal epithelial cells (Dunn and Wright 1987) and then migrate within the epithelium, continually invading and occupying the cytoplasm of new cells (Wright et al. 1987). To date, the mechanisms by which T. spiralis infective larvae recognize, invade, and migrate within the intestinal epithelium and establish their intramulticellular niche have not been elucidated. The larvae do not possess oral appendages or a spike (Bruce 1970), implying that the invasion of intestinal epithelial cells (IECs) may not be simply a result of mechanical penetration but may be mediated by surface glycoproteins and the oral secretions of the infective larvae (ManWarren et al. 1997). These larval proteins may interact with IECs and may play a key role during the larval invasion of IECs. However, the larval proteins related to invasion of IECs have not yet been identified (Nagano et al. 2009).
The use of conventional protein techniques to isolate Trichinella proteins has limited the discovery of new proteins that interact with host cells. Although an epithelial cell model for the in vitro invasion of Trichinella larvae has been developed (Gagliardo et al. 2002; ManWarren et al. 1997; Ren et al. 2011), the presence of contaminating enterocyte proteins in extracts of Trichinella larval protein makes it extremely difficult to obtain purified Trichinella proteins that interact with IECs. In addition, it is very difficult to determine the specific gene that encodes the Trichinella proteins that interact with host enterocytes. To overcome these limitations, phage display technology was used in this study to characterize these biomolecular interactions.
Since phage display technique was developed by Smith (1985), it has been used as a powerful technique for the selection of ligands that bind to any desired target (Paschke 2006; Xiao et al. 2007; Guo et al. 2010). In the phage display technique, peptides or proteins are expressed on the surface of phages as fusion proteins (Smith 1988). This allows the selection and amplification of phage clones with specific binding activities. Previous studies using Trichinella phage display libraries focused primarily on early diagnostic antigens (Zocevic et al. 2011). However, to the best of our knowledge, there has been no report of the use of the phage display technique to identify Trichinella proteins that bind to intestinal epithelial cells. In this study, we used a novel approach based on phage display libraries to screen for unidentified Trichinella proteins that interact with normal mouse IECs.
Materials and methods
Parasites and experiment animals
The isolates (ISS534) of T. spiralis used in this study were obtained from a domestic pig in Nanyang City of Henan Province, China. The isolate was maintained by serial passaging in Kunming mice in our laboratory. Six-week-old female BALB/c mice were obtained from the Experimental Animal Center of Henan Province (Zhengzhou, China). The mice were maintained under specific pathogen-free conditions with positive-pressure filtered air and sterilized food and water. All procedures of animal experiment were approved by the Life Science Ethics Committee of Zhengzhou University (Permission No. SYXK 2007-0009).
ML was recovered from infected mice at 42 days post infection by artificial digestion as described previously (Gamble et al. 2000; Li et al. 2010; Yang et al. 2010). After recovery, the larvae were washed three times with normal saline and then orally inoculated into 50 mice, with 5,000 ML per mouse in a volume of 100 μl. All the infected mice were euthanized by anesthetic inhalation of isoflurane (Sigma, USA) at 2 hpi. The small intestines were collected, cut along their entire length, and washed in pre-warmed phosphate-buffered saline (PBS) supplemented with antibiotics (200 U/ml penicillin and 200 μg/ml streptomycin). Then, the small intestines were cut into pieces and incubated in PBS at 37 °C for 2 h on a 300-μm mesh sieve. The released intestinal infective larvae were separated from intestinal debris by filtration through a 200-μm mesh sieve and differential sedimentation for 30 min (Ren et al. 2011). After several washes in sterile RNase-free water, the larvae were centrifuged at 600 × g for 10 min and stored at −80 °C until use.
Intestinal epithelial cells
In our experiments, normal mouse IECs used were obtained from fetal mouse small intestines and were susceptible to invasion by T. spiralis (Ren et al. 2011). The IECs were cultured (5 % CO2, 37 °C) in complete DMEM containing 4 mM glutamine, 20 mM Hepes, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 U/ml streptomycin, 0.1 U/ml bovine insulin (Sigma), and 10 % fetal bovine serum (Gibco). The cells were used at passage 8 for the experiment. Cell monolayers were dispersed by trypsinization (0.5 % trypsin–0.54 mM EDTA in PBS, at 23 °C for 5 min).
RNA isolation and construction of a Trichinella phage display library
Total RNA was isolated from the intestinal infective larvae of T. spiralis at 2 hpi using TRIzol reagent according to the manufacturer’s instructions (Invitrogen). The purified total RNA was quantified with a spectrophotometer (Nanodrop) at wavelengths of 230, 260, and 280 nm as described previously (Wang et al. 2008). The integrity of the total RNA was verified by running samples on 1.5 % agarose gels. Then, messenger RNA (mRNA) was purified from the total RNA using an Oligotex mRNA Mini kit (Qiagen). Random-primed first-strand cDNA was synthesized using 4 μg of Trichinella mRNA. After second-strand synthesis, the cDNA ends were modified using Novagen’s T7Select 10-3 Orient Express TM cDNA cloning system. Excess linkers and cDNAs shorter than 250 base pairs (bp) were removed by gel filtration. The Trichinella phage display library was created by inserting the modified cDNAs into the T7Select 10-3b EcoRI/HindIII vector arms. The recombinant vectors were subsequently packaged with 25 μl of T7 Packaging Extracts and propagated in Escherichia coli (strain BLT5403), during which time the fusion proteins were expressed and displayed on the phage surface. Finally, the titer of the packaged phage library was determined using a plaque assay, and the amplified library was stored at −80 °C in 10 % glycerol.
Biopanning
The T7 phage display cDNA library from T. spiralis intestinal infective larvae was screened by biopanning against normal mouse IECs. Cells grown to subconfluence (forming a monolayer) in a plastic 96-well ELISA plate were washed once with pre-warmed culture medium and further incubated for 30 min with culture medium containing 0.1 % bovine serum albumin (BSA). Phages were added to the 96-well culture plate to a final concentration of 2–3 × 108 plaque-forming unit (pfu)/ml, and the plate was incubated for 2 h at 37 °C. The cells were washed five times with PBS containing 0.05 % Tween-20 (PBST), and then the cell surface-bound phages were eluted for 20 min at room temperature by adding 200 μl of PBST containing 4 M urea. The obtained phages were amplified overnight at 37 °C in 50 ml of early log-phase E. coli BLT5403 cells. Following amplification, the lysed culture was centrifuged at 8,000 × g for 10 min, and the supernatant was used for the next round of biopanning. A total of five rounds of biopanning were performed. The same amount of phages (2 × 107 phages) was used for each round. After the fifth round of biopanning, the final eluted phages were tittered, and DNA was extracted from individual plaques by incubation in 100 μl of 10 mM EDTA (PH 8.0) at 65 °C for 10 min. The cDNA insert sizes were determined by PCR using 1 μl of phage lysate, 12.5 μl of PCR Master Mix (Roche), and 0.25 μl of T7Select-specific primers.
Sequence analysis
The T7 amplified PCR products were sequenced using an automated sequencer (Applied Biosystems). After removal of the flanking vector regions, the DNA sequences were compared with the GenBank database using the NCBI-BLAST server (http://www.ncbi.nlm.nih.gov/BLAST). The signatures of the proteins encoded by the Trichinella genes were queried against InterPro member databases by InterProScan searching (http://www.ebi.ac.uk/InterProScan/), and classifications were performed using Gene Ontology Annotation database (GOA; http://www.ebi.ac.uk/goa/) according to the protein accession numbers.
Phage binding assay
The positive phage clones, which were selected from the Trichinella library by biopanning, were individually examined for their binding to normal mouse IECs. Each positive phage clone was added to mouse IEC monolayer in a 96-well plate, and the input titers were 1 × 108, 1 × 107, 1 × 106, 1 × 105, and 1 × 104 pfu/ml. The cell surface-bound phages were eluted after incubation for 1 h. Other detailed procedures were the same as described above for “Biopanning.” Empty T7 phage and BSA were used as negative controls.
Results
RNA isolation and construction of a Trichinella phage display library
High-quality total RNA was isolated from T. spiralis intestinal infective larvae. The average RNA yield was 231.4 μg/g. The average A260/280 ratio ranged from 1.93 to 2.01, indicating a lack of protein contamination. The A260/230 ratio ranged from 2.11 to 2.32, suggesting that the RNA was of high purity and was not contaminated with polyphenol and polysaccharides. The RNA integrity was assessed based on the clarity of the ribosomal RNA bands visualized on a non-denaturing agarose gel, and distinct 28S, 18S, and 5S rRNA bands without degradation were observed (Fig. 1a).

RNA isolation and construction of a Trichinella phage display library. a Agarose gel electrophoresis of total RNA extracted from T. spiralis intestinal infective larvae. Lane M: RNA size markers (TaKaRa); lanes 1 and 2: total RNA from T. spiralis intestinal infective larvae. b Agarose gel electrophoresis of Trichinella cDNA. Lane M: DNA size markers (TaKaRa); lane 1: end-modified and purified cDNAs of T. spiralis infective larvae. To determine the size distribution of the Trichinella cDNAs, 3 μg of the cDNAs was processed for electrophoresis in 1.5 % agarose gels. The Trichinella cDNA appeared as a smear of fragments larger than 250 bp. c PCR amplification of phage clones randomly selected from the Trichinella phage display library. The PCR products of 32 phage clones chosen randomly from the primary library were analyzed by gel electrophoresis and visualized by ethidium bromide staining (lanes 1–32). DNA size markers (TaKaRa) are shown in lane M
After modification and purification, the Trichinella cDNA appeared as a smear of fragments larger than 250 bp (Fig. 1b). Based on the pfu after in vitro packaging, it was calculated that the T7 phage display library derived from T. spiralis larvae contained 5.4 × 106 independent clones. According to the Clarke–Carbon formula, a cDNA library should theoretically contain at least 3.3 × 105 independent clones to ensure that clones derived from low-abundance mRNAs would be present in the library with a 99 % probability (Sambrook and Russell 2001). Because the size of the Trichinella phage display library constructed in this experiment exceeds the statistically required number, it is highly likely that most of the Trichinella genes were represented in the library. The amplification of inserts in randomly selected clones revealed that their molecular weights ranged from 250 bp to 2.0 kb (Fig. 1c). Thirty-two inserted fragments were sequenced, and the results showed that 31 (97 %) of the 32 clones contained cDNA inserts that represent different Trichinella genes. These results indicated that the quality of the library was high.
Biopanning
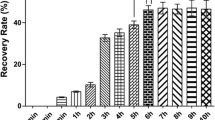
Five rounds of biopanning of the Trichinella cDNA phage display library with normal mouse IECs were performed. The recovery rate increased significantly in the first three rounds of biopanning, but no further enrichment was observed in the fourth and fifth rounds (Fig. 2). The phage titer of the eluted solutions increased from 105 pfu/ml (after the first round of biopanning) to 107 pfu/ml (after the fifth round of biopanning), indicating the successful enrichment of specifically bound phages.

Analysis of T7 phages eluted after each round of biopanning. The Trichinella phage display library was screened using a typical biopanning procedure with normal mouse intestinal epithelial cells (IECs). The phage titer in the eluates (before amplification) from each round was determined by a plaque assay
Sequence analysis of positive clones from the Trichinella phage display library
After the fifth round of biopanning, 50 positive plaques were randomly selected and amplified, and the cDNA inserts encoding the displayed peptides were sequenced. Ten unique genes were identified by screening the Trichinella phage display library with normal mouse IECs (Fig. 3a and Table 1). Clone Tsp1 showed high sequence identity with a previously identified T. spiralis gene encoding the calcium-transporting ATPase 2 protein, and clone Tsp2 aligned with the T. spiralis gene for hypothetical protein Tsp_03404. Clone Tsp3 showed high similarity to the T. spiralis gene encoding a conserved hypothetical protein (Tsp_03391), and clone Tsp4 was similar in sequence to a putative T. spiralis ovochymase-1 gene (Mitreva et al. 2011). The T. spiralis FACT complex subunit SPT16 was also identified (clone Tsp5). Clones Tsp6, Tsp7, Tsp8, and Tsp9 encoded polypeptides that showed sequence similarity to T-complex protein 1 subunit eta, glycosyl hydrolase family 47, DNA replication licensing factor MCM3, and the CBF/Mak21 family protein, respectively. Sequence analysis revealed that clone Tsp10 was identical to a putative T. spiralis nudix hydrolase gene identified previously (Wu et al. 2009).

Ten proteins that bind to normal mouse IECs were selected from the T. spiralis phage display library. a Agarose gel electrophoresis of positive phages selected from the Trichinella phage display library by biopanning on normal mouse IECs. After the fifth round of biopanning, 50 phage clones were randomly picked from individual plaques, and the cDNA inserts were amplified by PCR and sequenced. Sequence analysis showed that the 50 positive clones represented 10 unique genes. The ten different cDNA inserts were analyzed on a 1.5 % agarose gel. DNA size markers (TaKaRa) are shown in lane M. Lanes 1–10: the different cDNA inserts, which were named Tsp1-10. b Molecular function categories of the ten selected T. spiralis proteins that bind to normal mouse enterocytes according to their GO terms. Out of the ten proteins, six were annotated within molecular functions, and the pie chart shows the number of proteins in each GO category (level 2)
The classification of the ten identified proteins in terms of molecular function, biological process, and cellular localization was performed using the Gene Ontology Annotation database. Six proteins (Tsp1, calcium-transporting ATPase 2 protein; Tsp4, ovochymase-1; Tsp6, T-complex protein 1 subunit eta; Tsp7, glycosyl hydrolase family 47; Tsp8, DNA replication licensing factor MCM3; Tsp10, nudix hydrolase) of the T. spiralis proteins were annotated with putative molecular functions. Out of these six proteins, five have catalytic activity, four have binding activity, and one has transporter activity (Fig. 3b).
Phage binding assay
Within the range of 108–104 pfu/ml, the number of clone Tsp10 phages bound to mouse IECs (output) increased with the number of input phages, but there was no increase in the number of the control phages bound to IECs (Fig. 4). Similar results were obtained in the binding assays for the other nine clones (data not shown).

The specific assay for recombinant phage Tsp10 bound to normal mouse IECs. The number of Tsp10 phages bound to mouse IECs (output) increased with an increasing number of input phages. However, there was no significant change in the number of the control T7 phages that bound to mouse IECs
Discussion
In the present study, we used a T7 bacteriophage system to display Trichinella proteins as fusions with the capsid protein. The T7 display system was selected because of its advantages over the filamentous phage display system. Compared with filamentous phages, the T7 phage can display a wider variety of peptides or proteins on its surface. T7 phage particles assemble in the cytoplasm of E. coli cells, and progeny phages are released by cell lysis. Therefore, the displayed peptides do not need to be capable of secretion through the periplasm and the cell membrane, as required when using a filamentous phage (Russel 1991). Moreover, the T7 phage can grow faster and withstand harsh elution conditions that inactivate other types of phages; this characteristic makes the T7 phage an ideal choice for biopanning. The T7 phage display system has been widely used (Ishi and Sugawara 2008; Videlock et al. 2004). In this study, 5.4 × 106 independent clones were displayed on the surface of T7 phages using a cDNA library for T. spiralis infective larvae. The results indicated that most of the expressed genes should be represented in the T7 expression library that we constructed.
Although it has been known for many years that T. spiralis larvae invade the intestinal epithelium, the complex interactions between the parasite and host enterocytes are not yet fully understood (Suzuki et al. 2008). Previous studies on the tyvelose-specific antibody-mediated disruption of the intestinal niche have provided insight into the interactions between the parasite and host enterocytes (McVay et al. 2000). The earliest events in niche establishment by T. spiralis are likely to involve the recognition and invasion of the enterocytes by the parasite. The results of the in vitro experiments suggested that recognition and invasion required the active participation of both the larvae and the enterocytes (Butcher et al. 2000; ManWarren et al. 1997). Although some of the parameters of invasion have been established, the details of the molecular events in the process have not been fully elucidated. Our previous studies have demonstrated that several novel proteins are produced by the infective larvae after co-culture with IECs, and some of these proteins entered the enterocytes (Wang et al. 2011; Wang et al. 2012b). However, it has not been clarified which larval proteins can interact with host enterocytes and play critical roles during recognition and invasion by T. spiralis. Thus, for the first time, the phage display technique was employed in this study to screen for larval proteins with a high affinity for IECs. In addition, normal IECs were chosen as the target of the phages to more closely mimic the natural host’s intestinal environment and to screen for the larval proteins that bind to host IECs.
After five rounds of biopanning, the IEC-bound phages were significantly enriched by more than 290 times, indicating the successful display of Trichinella polypeptides on the surface of the T7 phage. The results of searching the GenBank database using the NCBI-BLAST server revealed ten different insertion sequences that mapped to predicted T. spiralis genes. Several of these inserts were found more than once, a result that was attributed to the significant degree of enrichment of each of these clones from the primary library. The binding of each positive clone with IECs was also confirmed in a phage binding assay, demonstrating that the in-frame Trichinella binding sequences selected by biopanning were highly specific for normal mouse IEC membranes.
The classification results of the ten Trichinella genes showed that six genes were annotated with putative molecular functions. Out of the six T. spiralis proteins encoded by these genes, five have catalytic activity and four have binding activity. Although their biological functions are not yet fully known, these proteins are candidates that might be related to the larval invasion of host enterocytes. Certainly, these proteins might be involved in the process of invasion (they might bind to important structural components of the enterocyte membrane or reorganize the enterocyte skeleton during invasion) and might be expressed on the exterior of the parasite and be available for interaction with the host cells. These proteins might also be internal proteins of the worm that coincidentally interact with IECs. This hypothesis needs to be verified in further experiments. Additionally, the expression levels of four genes (Tsp3, Tsp4, Tsp5, and Tsp10) have been confirmed to be significantly upregulated in T. spiralis larvae after their exposure to IECs using suppression subtractive hybridization (unpublished data). Three T. spiralis proteins (Tsp6, Tsp8, and Tsp10) have been found in IECs co-cultured with the intestinal infective larvae by Western blot analysis (Wang et al. 2012b). Moreover, anti-Tsp10 antibodies were able to recognize the native Tsp10 protein mainly localized to the stichosome of T. spiralis and prevented the in vitro partial invasion of IECs by infective larvae; the mice immunized with the recombinant phage T7-Tsp10 showed a 62.8 % reduction in adult worms and a 78.6 % reduction in muscle larvae following challenge with T. spiralis muscle larvae (Cui et al. 2013).
Conclusion
Trichinella phage display libraries provide a powerful and novel approach for identifying new Trichinella proteins that interact with the normal enterocyte membrane. Ten Trichinella proteins that bind to normal mouse IECs were selected from the phage library that was constructed in this study. Most of these annotated proteins had binding or catalytic activity, which might be related to the invasion of enterocytes by T. spiralis. However, other techniques such as RNA interference will be required to further investigate the functions of these genes to better understand the mechanism of the host–parasite interactions.
References
Bruce RG (1970) Structure of the esophagus of the infective juvenile and adult Trichinella spiralis. J Parasitol 56(3):540–549
Butcher BA, Gagliardo LF, ManWarren T, Appleton JA (2000) Larvae-induced plasma membrane wounds and glycoprotein deposition are insufficient for Trichinella spiralis invasion of epithelial cells. Mol Biochem Parasitol 107(2):207–218
Cui J, Wang ZQ, Kennedy MW (2006) The re-emergence of trichinellosis in China? Trends Parasitol 22(2):54–55. doi:10.1016/j.pt.2005.12.006
Cui J, Ren HJ, Liu RD, Wang L, Zhang ZF, Wang ZQ (2013) Phage-displayed specific polypeptide antigens induce significant protective immunity against Trichinella spiralis infection in BALB/c mice. Vaccine 31(8):1171–1178. doi:10.1016/j.vaccine.2012.12.070
Campbell WC (1983) Trichinella and Trichinosis. Plenum, New York
Dunn IJ, Wright KA (1987) The response of the intestinal epithelium in B10.A mice to infection with Trichinella spiralis. J Parasitol 73(4):712–722
Gagliardo LF, McVay CS, Appleton JA (2002) Molting, ecdysis, and reproduction of Trichinella spiralis are supported in vitro by intestinal epithelial cells. Infect Immun 70(4):1853–1859
Gamble HR, Bessonov AS, Cuperlovic K, Gajadhar AA, van Knapen F, Noeckler K, Schenone H, Zhu X (2000) International Commission on Trichinellosis: recommendations on methods for the control of Trichinella in domestic and wild animals intended for human consumption. Vet Parasitol 93(3–4):393–408
Guo A, Cai X, Jia W, Liu B, Zhang S, Wang P, Yan H, Luo X (2010) Mapping of Taenia solium TSOL18 antigenic epitopes by phage display library. Parasitol Res 106(5):1151–1157. doi:10.1007/s00436-010-1786-1
Ishi K, Sugawara F (2008) A facile method to screen inhibitors of protein–protein interactions including MDM2-p53 displayed on T7 phage. Biochem Pharmacol 75(9):1743–1750. doi:10.1016/j.bcp.2008.01.020
Kang SA, Cho MK, Park MK, Kim DH, Hong YC, Lee YS, Cha HJ, Ock MS, Yu HS (2012) Alteration of helper T-cell related cytokine production in splenocytes during Trichinella spiralis infection. Vet Parasitol 186(3–4):319–327. doi:10.1016/j.vetpar.2011.12.002
Li F, Cui J, Wang ZQ, Jiang P (2010) Sensitivity and optimization of artificial digestion in the inspection of meat for Trichinella spiralis. Foodborne Pathog Dis 7(8):879–885. doi:10.1089/fpd.2009.0445
ManWarren T, Gagliardo L, Geyer J, McVay C, Pearce-Kelling S, Appleton J (1997) Invasion of intestinal epithelia in vitro by the parasitic nematode Trichinella spiralis. Infect Immun 65(11):4806–4812
McVay CS, Bracken P, Gagliardo LF, Appleton J (2000) Antibodies to tyvelose exhibit multiple modes of interference with the epithelial niche of Trichinella spiralis. Infect Immun 68(4):1912–1918
Mitreva M, Jasmer DP, Zarlenga DS, Wang Z, Abubucker S, Martin J, Taylor CM et al (2011) The draft genome of the parasitic nematode Trichinella spiralis. Nat Genet 43(3):228–235. doi:10.1038/ng.769
Nagano I, Wu Z, Takahashi Y (2009) Functional genes and proteins of Trichinella spp. Parasitol Res 104(2):197–207. doi:10.1007/s00436-008-1248-1
Paschke M (2006) Phage display systems and their applications. Appl Microbiol Biotechnol 70(1):2–11. doi:10.1007/s00253-005-0270-9
Pozio E (2007) World distribution of Trichinella spp. infections in animals and humans. Vet Parasitol 149(1–2):3–21. doi:10.1016/j.vetpar.2007.07.002
Ren HJ, Cui J, Wang ZQ, Liu RD (2011) Normal mouse intestinal epithelial cells as a model for the in vitro invasion of Trichinella spiralis infective larvae. PLoS One 6(10):e27010. doi:10.1371/journal.pone.0027010
Russel M (1991) Filamentous phage assembly. Mol Microbiol 5(7):1607–1613
Sambrook J, Russell D (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Lab, New York
Smith GP (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228(4705):1315–1317
Smith GP (1988) Filamentous phages as cloning vectors. Biotechnology 10:61–83
Suzuki T, Sasaki T, Takagi H, Sato K, Ueda K (2008) The effectors responsible for gastrointestinal nematode parasites, Trichinella spiralis, expulsion in rats. Parasitol Res 103(6):1289–1295. doi:10.1007/s00436-008-1130-1
Videlock EJ, Chung VK, Mohan MA, Strok TM, Austin DJ (2004) Two-dimensional diversity: screening human cDNA phage display libraries with a random diversity probe for the display cloning of phosphotyrosine binding domains. J Am Chem Soc 126(12):3730–3731. doi:10.1021/ja039006p
Wang X, Tian W, Li Y (2008) Development of an efficient protocol of RNA isolation from recalcitrant tree tissues. Mol Biotechnol 38(1):57–64. doi:10.1007/s12033-007-0073-6
Wang SW, Wang ZQ, Cui J (2011) Protein change of intestinal epithelial cells induced in vitro by Trichinella spiralis infective larvae. Parasitol Res 108(3):593–599. doi:10.1007/s00436-010-2102-9
Wang ZQ, Li LZ, Jiang P, Liu LN, Cui J (2012a) Molecular identification and phylogenetic analysis of Trichinella isolates from different provinces in mainland China. Parasitol Res 110(2):753–757. doi:10.1007/s00436-011-2549-3
Wang ZQ, Wang L, Cui J (2012b) Proteomic analysis of Trichinella spiralis proteins in intestinal epithelial cells after culture with their larvae by shotgun LC-MS/MS approach. J Proteomics 75(8):2375–2383. doi:10.1016/j.jprot.2012.02.005
Wright KA, Weidman E, Hong H (1987) The distribution of cells killed by Trichinella spiralis in the mucosal epithelium of two strains of mice. J Parasitol 73(5):935–939
Wu XP, Fu BQ, Wang XL, Yu L, Yu SY, Deng HK, Liu XY, Boireau P, Wang F, Liu MY (2009) Identification of antigenic genes in Trichinella spiralis by immunoscreening of cDNA libraries. Vet Parasitol 159(3–4):272–275. doi:10.1016/j.vetpar.2008.10.035
Xiao Y, Zhou Y, Wang J, Yu M, Wang G, Jin J, Xiao J (2007) Selection and identification of human gonadotropin-releasing hormone promoter binding peptides by phage display-CEMSA. J Mol Recognit: JMR 20(1):51–57. doi:10.1002/jmr.811
Yang Y, Jian W, Qin W (2010) Molecular cloning and phylogenetic analysis of small GTPase protein Tscdc42 from Trichinella spiralis. Parasitol Res 106(4):801–808. doi:10.1007/s00436-010-1735-z
Zocevic A, Mace P, Vallee I, Blaga R, Liu M, Lacour SA, Boireau P (2011) Identification of Trichinella spiralis early antigens at the pre-adult and adult stages. Parasitology 138(4):463–471. doi:10.1017/S0031182010001526
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81271860, 30972579, and 30972492).
Author information
Authors and Affiliations
Corresponding authors
Additional information
The nucleotide sequence data reported in this paper are available in the GenBank™ database under the accession numbers JX478226, JX478227, JX478228, JX478229, JX478230, JX478231, JX478232, JX478233, JX478234, and JX478235.
Rights and permissions
About this article
Cite this article
Ren, H.J., Liu, R.D., Wang, Z.Q. et al. Construction and use of a Trichinella spiralis phage display library to identify the interactions between parasite and host enterocytes. Parasitol Res 112, 1857–1863 (2013). https://doi.org/10.1007/s00436-013-3339-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-013-3339-x