
Abstract
Avian malaria is of significant ecological importance and serves as a model system to study broad patterns of host switching and host specificity. The erythrocyte invasion mechanism of the malaria parasite Plasmodium is mediated, in large part, by proteins of the erythrocyte-binding-like (ebl) family of genes. However, little is known about how these genes are conserved across different species of Plasmodium, especially those that infect birds. Using bioinformatical methods in conjunction with polymerase chain reaction (PCR) and genetic sequencing, we identified and annotated one member of the ebl family, merozoite apical erythrocyte-binding ligand (maebl), from the chicken parasite Plasmodium gallinaceum. We then detected the expression of maebl in P. gallinaceum by PCR analysis of cDNA isolated from the blood of infected chickens. We found that maebl is a conserved orthologous gene in avian, mammalian, and rodent Plasmodium species. The duplicate extracellular binding domains of MAEBL, responsible for erythrocyte binding, are the most conserved regions. Our combined data corroborate the conservation of maebl throughout the Plasmodium genus and may help elucidate the mechanisms of erythrocyte invasion in P. gallinaceum and the host specificity of Plasmodium parasites.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Malaria is caused by the parasite genus Plasmodium and undergoes three major life stages: a sexual stage within the mosquito’s midgut, the generally asymptomatic exoerythrocytic stage, and the symptomatic blood stage in the vertebrate host, which is characterized by merozoite invasion of erythrocytes. This process is mediated by receptor/ligand interactions that can vary depending on the species of Plasmodium or host polymorphisms in genes encoding the receptors (Chitnis and Blackman 2000; Ghai et al. 2002). For example, Plasmodium vivax requires the Duffy-binding protein to recognize Duffy blood group antigens on the surface of human erythrocytes (Wertheimer and Barnwell 1989). In contrast, Plasmodium falciparum has several proteins that recognize receptors on human erythrocytes (Brown and Higgins 2010). A major group of these receptors are part of the family of erythrocyte-binding-like (EBL) proteins that includes EBA-175, a P. falciparum protein that binds to glycophorin A on human erythrocytes and plays an important role in host specificity and the junction formation involved in erythrocyte invasion (Orlandi et al. 1992; Tolia et al. 2005). Other members of the EBL family in P. falciparum include JESEBL, PEBL, EBL-1, BAEBL, and MAEBL (Adams et al. 1992).
Merozoite apical erythrocyte-binding ligand (MAEBL) has been described in Plasmodium species that infect mammals and rodents (Blair et al. 2002; Michon et al. 2002; Verra et al. 2006b). MAEBL is a protein of the EBL family with characteristics that distinguish it from other family members (Blair et al. 2002). All ebl genes are single copy and share similar gene structures: conserved exon–intron boundaries and conserved amino and carboxyl cysteine-rich domains (Adams et al. 2001). In addition, most ebl genes encode duplicate cysteine-rich Duffy-binding-like ligand domains; however, MAEBL does not. Instead, MAEBL has duplicate amino cysteine-rich regions (M1 and M2) with similarity to domains I and II of apical membrane antigen-1 (AMA-1) (Kappe et al. 1998). The MAEBL M1 and M2 domains are responsible for the recognition of a presumed specific receptor (Ghai et al. 2002). Both domains are conserved, but only the M2 MAEBL domain appears to be essential for binding its receptor on erythrocytes (Ghai et al. 2002). The unknown MAEBL receptor is not a sialylated protein but is affected by both papain and trypsin digestion (Ghai et al. 2002). Unlike its EBP counterparts, which are expressed in micronemes, MAEBL is expressed in rhoptries, apical organelles that secrete proteins during merozoite invasion. Furthermore, MAEBL is detected in late trophozoite and early schizont stages prior to erythrocyte rupture and Duffy-binding protein expression in the rodent parasite Plasmodium yoelli (Noe and Adams 1998).
MAEBL has been promoted as a target for vaccine production because of its expression in both merozoite and sporozoite stages within the host (Ghai et al. 2002; Preiser et al. 2004). Maebl is alternatively spliced, producing different transcripts at the sporozoite and merozoite stages (Singh et al. 2004). It has also been shown that the maebl transcript codes for a bicistronic message, containing the mitochondrial ATP synthase subunit gene upstream of the transcript (Balu et al. 2009). For these reasons, maebl serves as a model for post-transcriptional mRNA processing of Apicomplexans. Additionally, MAEBL plays an important role in many developmental stages, both in the host and mosquito vector (Florens et al. 2002). The protein has been detected in zygote, ookinete, merozoites, midgut, and salivary sporozoites, indicating that it is present at various developmental stages and remains important throughout the life of the parasite (Blair et al. 2002; Ghai et al. 2002; Patra et al. 2008; Preiser et al. 2004; Saenz et al. 2008; Singh et al. 2004).
The maebl gene has been identified in P. falciparum isolates and has high conservation with orthologs of rodent malaria parasites (Blair et al. 2002). Among both laboratory clones and field isolates, maebl shows conservation with few polymorphisms in the M1 and M2 extracellular binding domains (Verra et al. 2006a). As of yet, maebl has not been characterized in an avian malaria parasite. Avian Plasmodium spp. are numerous, are evolutionary basal to mammalian Plasmodium spp., and provide a natural model system to study the ecology and evolution of malaria parasites (Martinsen et al. 2008; Ricklefs and Outlaw 2010). The gene is of ancient origin and appears to have evolved as a single locus prior to Plasmodium speciation (Michon et al. 2002). The isolation of a functional maebl gene in an avian Plasmodium species may aid in deciphering the evolution of erythrocyte invasion processes of the Plasmodium genus. Thus, pending identification of maebl in an avian species, we hypothesized that there would be a close phylogenetic relationship of the avian malaria maebl to its mammalian orthologs. Here, we sought to identify the sequence and expression of maebl in the chicken parasite, Plasmodium gallinaceum, the only avian malaria parasite with a published genome (Wellcome Trust Sanger Institute) (Silva et al. 2011). Our statistical and molecular data show that the sequence and gene structure of P. gallinaceum maebl is orthologous to other Plasmodium species infecting mammals. Results confirm a high level of amino acid conservation (based on synonymous and non-synonymous changes) among P. gallinaceum maebl and human Plasmodium species, lending evidence to the biological importance of the gene. In addition, we confirm the expression of maebl in P. gallinaceum and use DNA sequencing to annotate the gene.
Materials and methods
Identification and sequence analysis of the full length P. gallinaceum maebl gene
BLAST searches of the P. gallinaceum genome sequence project (accession number PRJNA12649) carried out with the full-length P. falciparum EBL genes eba-175, jesebl, pebl, baebl, and ebl-1 (Adams et al. 2001) yielded no significant hits. Only one BLAST search of the P. gallinaceum genome sequence project (accession number PRJNA12649) carried out with the full-length P. falciparum MAEBL sequence (accession number XM_001348117) as query (Altschul et al. 1990) was significant. We also performed local BLAST analysis using the same P. gallinaceum FTP genome database (PRJNA12649; Parasite Genomics Group at the Wellcome Trust Sanger Institute) that yielded nearly identical results.
All BLAST searches, using P. falciparum as query, identified gal28as.d000006237.Contig1 (a super phusion contig) to contain the P. gallinaceum maebl ortholog sequence. The downloaded super phusion contig was originally 10,549 bp and contained an unknown sequence, which was labeled N10 by the Parasite Genomics Group. Primers flanking the N10 sequence were designed in order to obtain the missing sequence (Fig. S1) and confirm the downloaded sequence. The unknown N10 sequence was 235 bp, making the super phusion contig 10,774 bp long. The complete super phusion contig (gal28as.d000006237.Contig1) was recorded and translated to identify open reading frames using Sequencher 4.8 (GeneCodes, Ann Arbor, MI, USA) and Apollo (Lewis et al. 2002).
Annotations
Multiple sequences obtained from GenBank, included EST and proteomic sequences, were uploaded onto the automated genome annotation pipeline MAKER (Cantarel et al. 2008) to annotate and create a model of the P. gallinaceum MAEBL gene region. Annotations were viewed in Apollo (Lewis et al. 2002) and were manually adjusted so that they were consistent with maebl orthologs of various Plasmodium species.
Phylogenetic analysis
We used Bayesian analysis to construct a phylogenetic tree using the M2 domain sequences (of seven Plasmodium spp.: P. falciparum, P. gallinaceum, P. yoelli, P. vivax, Plasmodium knowlesi, Plasmodium chabaudi, and Plasmodium berghei (GenBank accession numbers XM_001348117, JQ780838, XM_724578, AY042083, XM_002259441, and plasmoDB.org identifiers PCHAS_070250 and PBANKA_090130, respectively). All individual sequences were grouped into a consensus that was 1,231 bp long, with P. falciparum (XM_001348117) as the outgroup. Data were generated by MrBayes version 3.1.2 (Huelsenbeck et al. 2001), using the model of sequence evolution obtained from MrModeltest: Generalized Time Reversible, Gamma and Proportion Invariant (GTR + I + G). Two Markov chains were run simultaneously for ten million generations and sampled every 200 generations, producing 50,000 trees. Twenty-five percent of the trees were discarded, and the remaining 37,500 trees were used to construct a majority consensus tree.
Gene/protein alignments and divergence levels
Gene and protein sequences were aligned using ClustalX (Larkin et al. 2007), and rates of synonymous and non-synonymous changes ((K a/K s ratios) were measured with the PAL2NAL package (Suyama et al. 2006).
DNA/RNA extractions, PCR, and sequencing
Genomic DNA from White Leghorn chicken blood infected with P. gallinaceum strain A8 was used for all molecular work (Frevert et al. 2008). The blood was provided by Dr. Ute Frevert. Parasite DNA was extracted from blood following the animal tissue protocol of the Wizard® SV Genomic DNA Purification Kit (Promega Corporation, Madison, WI, USA). All polymerase chain reactions (PCR) involving genomic DNA were carried out in a 25-μl reaction mixture containing 10–100 ng of genomic DNA (1 μl of template DNA), 0.4 mM of each dNTP, 0.4 mM of each primer, 2.5 μl of 5X CL Buffer (Qiagen, Valencia, CA, USA), 0.625 U Taq (Qiagen, Valencia, CA, USA), and 0.4 mg/ml bovine serum albumin. Several primer combinations were used to amplify smaller fragments of maebl (Table S1). Positive PCR products were purified using ExoSap (following the manufacturer’s instructions, USB Corporation, Cleveland, OH, USA), then sent out for Bi-directional sequencing to Elim Biopharmaceuticals Inc., Hayward, CA, USA. All PCR reactions were tested in duplicates, to verify all sequence data. The GenBank accession number for the P. gallinaceum 6,866-bp maebl gene is JQ780838. These data have also been uploaded to PlasmoDB.org.
To obtain total RNA, we infected chickens with P. gallinaceum. Six White Leghorn chickens were hatched and raised at UC Davis. After 6 days, five of the chickens were infected by intraperitoneal injection of infected brain emulsions, again provided by Dr. Frevert (Frevert et al. 2008). One chicken was left uninfected to serve as a negative control. Infected blood was drawn 7 days after infection and stored in TRIzol® LS Reagent (Invitrogen, Grand Island, NY, USA) and processed at San Francisco State University. Blood was drawn a second time at day 15 and third time at day 21 post-infection. Infection status was confirmed visually both by microscopy, after staining blood smears with Giemsa, and using a standard PCR protocol to amplify the Cytochrome b gene (Valkiūnas et al. 2009). RNA was extracted from the second bleed using the TRIzol® LS Reagent (Invitrogen, Grand Island, NY, USA) protocol with slight modifications. Heavy phase-lock gel tubes were used to separate nucleic acids into an aqueous phase. RNA was precipitated using isopropyl alcohol and a 250 μl of a high-salt solution (0.8 M sodium citrate, 0.2 M NaCl in diethylpyrocarbonate (DEPC)-treated water). The RNA was resuspended and treated with Ambion® TurboDNase™. RNA was re-extracted with phenol-chloroform isoamyl and precipitated in 100 % ethanol. The RNA pellet was resuspended in DEPC-treated water. cDNA was synthesized via reverse transcriptase PCR using 2 μg of pure RNA from infected chickens using an iScript™ cDNA synthesis kit (BIORAD Laboratories).
All PCR reactions using cDNA as template were carried out using AccuPower® HotStart PCR PreMix (Bioneer, Alameda, CA, USA). Primers were mixed with sterile water and added to the PCR tubes to make a total volume of 20 μl including the cDNA template. The final concentration of each primer was 0.75 μM.
To confirm cDNA synthesis, chicken α-globin genes were amplified with primer sets AlphaA (αA) and AlphaD (αD) (Rincon-Arano et al. 2009). PCR was conducted using thermal cycling profiles described by the authors. Sequencing was carried out in the same manner as for genomic DNA.
Maebl expression was detected by using a nested PCR protocol. For the first PCR, we amplified a 736-bp fragment using primers A12_F5: 5′-GAAGATCGACTAAATGATCCAACAA-3′ and M2A_R3: 5′-CTAAGCAAGGTCCTACACAGTATG-3′. Initial denaturation was 94 °C for 3 min, followed by 20 cycles of 94 °C for 30 s, 58 °C for 30 s, 72 °C for 1 min, and a final extension at 72 °C for 10 min. The second PCR amplified a 199-bp fragment using primers A13_F5: 5′-GATTGCCATGCATCATGGTG-3′ and A12_R3: 5′-ACCTAATCTCCCACCTAAACC-3′. Initial denaturation was 94 °C for 3 min, followed by 35 cycles of 94 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s, and a final extension at 72 °C for 7 min. Expression was visualized on an agarose gel.
To confirm splice sites, we performed a nested PCR. The first PCR used 3 μl of cDNA and primers complimentary to regions in predicted exons 2 and 5: A10_F5: 5′-GCTTCTGGAAAACCAACACC-3′ and E4A_R3: 5′-AAGTTTACACATTAGCAATGATATG-3′, respectively. Initial denaturation was 94 ° C for 3 min, followed by 35 cycles of 94 °C for 30 s, 58 °C for 30 s, 72 °C for 1 min, and a final extension at 72 °C for 10 min. The second PCR used 3 μl of the first PCR product and primers complimentary to regions in predicted exons 3 and 4: E2B_F5: 5′-TGTTCTAATGAAGAAAGAGAACAT-3′ and A9_R3: 5′-TGGAGCAGGAATAGCACT-GAT-3, respectively. The thermal cycling profile was the same as the first PCR. The final 189-bp product was sequenced and further enhanced our maebl gene structure model (Fig. S2).
Results
Identification and conservation of maebl in P. gallinaceum
We identified an ortholog of maebl in the avian parasite, P. gallinaceum in the Wellcome Trust Sanger Institute database, and confirmed the sequence using PCR and genomic sequencing. The resulting sequence is approximately 6,866 bp (including introns) (GenBank accession number JQ780838). Although we performed DNA sequencing to confirm the majority of the gene, we were unable to sequence a 1,491-bp region due to high repeat density (positions 7278–8771), which codes for the repetitive region of MAEBL (Fig. 1c). Other groups studying mammalian Plasmodium species have also had difficulty obtaining sequence data for this region (Michon et al. 2002).

Schematic gene structure of Plasmodium MAEBL. a The conserved gene structure of maebl (above) in known Plasmodium species consists of five exons, coding (1) signal peptide; (2) ligands domains M1 and M2, repetitive region, and carboxyl-cysteine rich domain; (3) transmembrane domain; and a (4/5) cytoplasmic domain. b Apollo model of P. gallinaceum maebl based on EST, BLAST, and Protein2protein evidence (bars in black box). P. gallinaceum maebl gene structure appears conserved with five exons. P. gallinaceum transcript maebl model is indicated in the light gray box. c The gene structure of P. gallinaceum maebl as predicted by Apollo and Maker. The carboxyl-cysteine-rich domain and transmembrane domain are located in different exons than the conserved Plasmodium species gene structure, a
DNA sequences were translated and the resulting amino acid sequences showed 45 % amino acid identity to P. falciparum MAEBL and 63 % DNA identity (Fig. S3, S4). Like all the MAEBL proteins characterized so far, the most conserved regions are the cysteine-rich domains M1 and M2 (Michon et al. 2002). We found that P. gallinaceum maebl M1 and M2 domains have 16 cysteine residues that are conserved when compared to 14 other Plasmodium species (Fig. 2). Additionally, P. gallinaceum MAEBLs M1/M2 domains also share similarity to AMA-1 domains I and II with only ten of the 16 cysteines conserved (Ghai et al. 2002). The remainder of the P. gallinaceum maebl amino acid sequence still had a high degree of similarity to other MAEBL proteins.

P. gallinaceum M1 and M2 domains have conserved cysteine residues. CLUSTAL alignment of the P. gallinaceum MAEBL M1 and M2 domains. The 16 conserved cysteines are numbered. Black-highlighted regions represent conserved/identical amino acids. Gray regions represent similar amino acids. Spaces have been introduced to maximize alignment
Gene structure of P. gallinaceum maebl
The P. gallinaceum maebl gene contains an amino signal peptide, extracellular binding domains (M1/M2), a transmembrane domain, and a cytoplasmic tail (Fig. 1c). Using annotation evidence from MAKER, we were able to predict the exon–intron boundaries from P. gallinaceum maebl and show five exons by using expression data (ESTs) from Plasmodium spp. We also performed a nested PCR to determine the intron–exon boundary between predicted exons 3 and 4. The sequence obtained was sequenced and enhanced our maebl gene structure model (Fig. 1b). The M1 and M2 domains are coded at the end of exon 1 and continue through the first half of exon two. The rest of exon 2 codes for the tandem repeat region of MAEBL, containing mainly lysine, alanine, glutamic acid, and arginine residues. The carboxyl-cysteine domain homologous to region VI of the ebl family (Adams et al. 1992), the transmembrane domain, and the cytoplasmic tail are all shifted relative to the known maebl genes and are instead coded in exons 3, 4, and 5 (Fig. 1a). The total lengths of the exons differ between P. gallinaceum and P. falciparum (1,809 and 2,055 bp, respectively) and result from different lengths of the repeat region.
Phylogenetic analysis of M2 domain
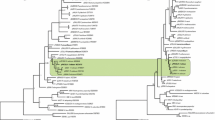
Based on the identified sequence of P. gallinaceum MAEBL, using the M2 domains of seven Plasmodium species, we constructed a maximum-likelihood tree (Fig. 3). The maebl gene from P. gallinaceum is significantly divergent from that of the mammalian parasites P. knowlesi, P. vivax, P. berghei, P. yoelli, and P. chabaudi. However, the sequence data did not provide enough resolution to establish an informative relationship with P. falciparum.

P. gallinaceum MAEBL M2 domain diverges from rodent and mammalian Plasmodium spp. Phylogenetic relationships among the M2 domains of maebl from P. gallinaceum and mammalian Plasmodium species. Numbers along branches correspond to node support from Bayesian analysis
Molecular variation of the M2 domains of Plasmodium species compared to P. gallinaceum
Sequence variation of P. gallinaceum M2 maebl domain was calculated by counting non-synonymous and synonymous substitutions compared to other Plasmodium M2 MAEBL domains (Table 1). Maebl sequences were aligned and the K a/K s ratios were calculated. The rate of non-synonymous (K a) changes were all less than 0.1023, and the rate of synonymous (K s) changes was high, suggesting that purifying selection is acting on the M2 domain.
Maebl is expressed by P. gallinaceum
After identifying maebl within the genome of P. gallinaceum, we verified its expression. Following the synthesis of cDNA from the blood of an infected chicken, we used PCR with maebl-specific primers within the M2 domain to detect the expression of the gene. Results showed that a 199-bp fragment from maebl was present and that P. gallinaceum indeed expresses maebl in the blood stages (Fig. 4).

Expression of maebl by P. gallinaceum. Lane 1 (left) shows amplification of a 199-bp fragment amplified from cDNA using a nested PCR with maebl primers. Reverse transcriptase PCR mastermix without RNA and water were used as the template for the negative control lanes. αA primers and αD primers were used to amplify expressed chicken alpha-globin indicating the presence of cDNA
Discussion
Malaria parasites have evolved to infect a wide variety of hosts including mammals, reptiles, and birds. It is known that within each host, proteins expressed by the parasite mediate erythrocyte invasion. In this study, we investigated the ebl gene known as maebl in P. gallinaceum. Using bioinformatics, we were able to identify maebl, annotate it, and show its conservation across different species of Plasmodium. In addition, we verified that P. gallinaceum expresses maebl.
The 16 cysteine residues in each M1 and M2 domain of the P. gallinaceum maebl are conserved, which suggests that the residues may be essential in the formation of the protein’s tertiary structure, specifically in the extracellular binding domains. Comparison of the AMA-1 crystal structure to its paralog MAEBL reveals that the ten conserved cysteines form similar disulfide bridges, but not enough information was present to predict the structure of the six remaining cysteine residues and their possible functions (Chesne-Seck et al. 2005). Thus, determining MAEBL’s crystal structure will confirm the importance and function of the conserved cysteine residues.
All five exons and exon–intron boundaries are conserved among P. falciparum, P. knowlesi, P. chabaudi, P. yoelli, P. vivax, and P. berghei (Fig. 1a), but the last three 3′ end exons have different exon–intron boundaries in P. gallinaceum (Fig. 1c). The conserved carboxyl-cysteine-rich region is coded in exon 3 of P. gallinaceum MAEBL instead of exon 2, further validating the distant relationship of P. gallinaceum compared to other mammalian Plasmodium species. Although the repetitive region cannot be confirmed, the fact that our cDNA primers (in a nested PCR reaction) were able to amplify a cDNA region downstream suggests that the predicted exons 2 and 5 exist in the cDNA.
The constructed M2 MAEBL phylogenetic tree is comparable to the known phylogenetic relationships of Plasmodium species and reveals the divergence of the M2 domain between P. gallinaceum and mammalian Plasmodium spp. (Martinsen et al. 2008; Outlaw and Ricklefs 2011). We focused on the M2 domain because studies show it is essential for erythrocyte binding when compared to the M1 domain (Ghai et al. 2002). The M2 domain of P. gallinaceum MAEBL showed no evidence of diversifying or positive selection based on the high rate of synonymous changes compared to the rate of non-synonymous substitutions. A ratio below 0.25 is indicative of purifying selection, and the quantification of evolution upon the P. gallinaceum M2 MAEBL domain shows K a/K s ratios below 0.1023 (Table 1). These calculated ratios strongly suggest that the M2 domain is functionally constrained and important as previously described (Verra et al. 2006b). Our analysis of substitution rates in P. gallinaceum M2 domain adds to Verra’s study and confirms the functional constraints on the M2 domain. Thus, although we find that the M2 domain differs at the nucleotide level from the same region of mammalian parasites (Fig. 3), we find that the domain is highly functionally conserved in all tested malaria parasites. The importance of the M2 domain in avian Plasmodium species is unknown, but it would be interesting to further study the M2 domain’s role in erythrocyte invasion in birds and also in sporogony in mosquitoes.
Studies have found that MAEBL is alternatively spliced in different Plasmodium stages (Ghai et al. 2002; Preiser et al. 2004; Singh et al. 2004). The MAEBL isoform containing the transmembrane domain is important in the invasion of the mosquito salivary glands and for erythrocyte binding in merozoites (Noe and Adams 1998; Saenz et al. 2008). MAEBL exemplifies a complex gene structure in Plasmodium because of its multiple isoforms and possible bicistronic transcript. These post-transcriptional processes appear to be conserved in the chicken parasite, and with the identification of maebl in P. gallinaceum, future studies can ascertain whether higher-level transcript regulation is occurring in avian Plasmodium spp.
The complex structure of MAEBL suggests that different receptors may be present in the host and vector. The high conservation observed in MAEBL may indicate the essential function of its receptor within the host. Thus, identification of the MAEBL receptor and further study of avian nucleated RBCs will lead to understanding the purpose and mechanisms of MAEBL. In a study by Fu et al. (2005), it was shown that P. falciparum survives without the protein, but the loss of MAEBL in merozoites alters the parasite invasion pathway to a novel alternative pathway, which is sialic acid-dependent and trypsin-insensitive. We can only speculate that P. gallinaceum may use this or a similar invasion pathway. The receptors of EBL proteins in mammalian Plasmodium species are mostly sialylated proteins such as glycophorins (Iyer et al. 2007; Mayer et al. 2009). Glycophorins from nucleated chicken erythrocytes have been isolated and characterized. It was found that they are highly divergent from their human counterparts, and therefore, the ebl genes of different Plasmodium spp. may differ as well (Duk et al. 2000). Although MAEBL is highly conserved, MAEBL from rodent Plasmodium spp. cannot bind to human RBCs and vice versa (Kappe et al. 1998). Crosnier et al. (2011) was able to identify the human erythrocyte receptor of PfRh5 (an essential merozoite erythrocyte-binding protein of P. falciparum) using a human erythrocyte protein library for an avidity-based extracellular interaction screen assay. A similar approach could potentially lead to the identification of the MAEBL receptor(s) in different hosts.
The identification of Plasmodium proteins involved in erythrocyte invasion is important in understanding the mechanism by which Plasmodium infects its host. For example, infected avian erythrocytes with P. gallinaceum develop furrow-like structures instead of knob-like structures like most Plasmodium spp. (Nagao et al. 2008). Avian parasites encounter different immunological pressures from their hosts, which result in different methods of erythrocyte invasion. In addition, erythrocyte-binding proteins are involved in the junction formation that commits the parasite to invade red blood cells. Thus, these proteins are being added to the list of possible vaccine targets against malaria because of their importance in the invasion mechanism (Drummond and Peterson 2005).
We focused on P. gallinaceum because it is the only avian Plasmodium to have had its genome sequenced. Our analysis using annotated Plasmodium ebl sequences, as well as using EBL Duffy-binding domains, did not detect any other ebl members. One possible reason may be that other ebl genes in P. gallinaceum are too divergent from mammalian parasites or that other ebl genes found in P. falciparum are not present in P. gallinaceum. For example, P. vivax has only one ebl gene, the Duffy-binding protein (Wertheimer and Barnwell 1989). The complete annotation of P. gallinaceum’s genome will reveal whether other ebl members or proteins involved in erythrocyte invasion are present and functional. To obtain this information, high throughput sequencing of the parasite’s transcriptome would confirm or refute the expression of other ebl genes in the parasite.
Avian malaria provides an ideal model system to test whether EBL proteins are involved in the host specificity of Plasmodium species. The extinction of several avian populations in the Hawaiian Islands was in part due to a newly introduced Plasmodium strain (Plasmodium relictum) that was capable of infecting multiple naïve bird species (a generalist parasite) (Beadell et al. 2009; Loiseau et al. 2010; Vanriper et al. 1986). Unlike generalist parasites, host-specific parasites (specialists) can infect only one species, and it is evident that certain malaria parasites are specialists to certain bird species (Iezhova et al. 2005; Loiseau et al. 2012). Moreover, previous work has shown that specialists may become generalists in host switching events, with increased virulence (Garamszegi 2006). Although the molecular basis of host specificity in avian Plasmodium parasites is largely unstudied, it is known that EBA-175 is a protein potentially involved in the host-specificity of P. falciparum and Plasmodium reichenowi of humans and chimpanzees, respectively (Chattopadhyay et al. 2006; Martin et al. 2005; Tolia et al. 2005). EBA-175 is highly conserved between P. falciparum and P. reichenowi, but neither can infect each other’s host. Chattopadhyay et al. (2006) found that by comparing the 3D structure of EBA-175 of both species, the only significant difference was the distribution of protein surface charge in the channel containing the duplicated Duffy-binding domains. Here we have shown that maebl, another member of the ebl family, is also conserved and expressed across various species of Plasmodium. Thus, the identification and functional characterization of ebl genes in avian parasites will aid in the prediction of potential emerging diseases in avian populations and help researchers to better understand the emergence of novel strains of malaria in both birds and other animals across the globe.
Abbreviations
- MAEBL:
-
Merozoite apical erythrocyte-binding ligand
- EBA-175:
-
Erythrocyte-binding antigen-175
- DBP:
-
Duffy-binding protein
- EBL:
-
Erythrocyte binding-like
- PCR:
-
Polymerase chain reaction
- RBC:
-
Red blood cells
- EST:
-
Expressed sequence tags
References
Adams JH, Blair PL, Kaneko O, Peterson DS (2001) An expanding ebl family of Plasmodium falciparum. Trends Parasitol 17:297–299
Adams JH, Sim BK, Dolan SA, Fang X, Kaslow DC, Miller LH (1992) A family of erythrocyte binding proteins of malaria parasites. Proc Natl Acad Sci USA 89:7085–7089
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Balu B, Blair PL, Adams JH (2009) Identification of the transcription initiation site reveals a novel transcript structure for Plasmodium falciparum maebl. Exp Parasitol 121:110–114
Beadell JS, Covas R, Gebhard C, Ishtiaq F, Melo M, Schmidt BK, Perkins SL, Graves GR, Fleischer RC (2009) Host associations and evolutionary relationships of avian blood parasites from West Africa. Int J Parasitol 39:257–266
Blair PL, Kappe SH, Maciel JE, Balu B, Adams JH (2002) Plasmodium falciparum MAEBL is a unique member of the ebl family. Mol Biochem Parasitol 122:35–44
Brown A, Higgins MK (2010) Carbohydrate binding molecules in malaria pathology. Curr Opin Struct Biol 20:560–566
Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Sanchez Alvarado A, Yandell M (2008) MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18:188–196
Chattopadhyay D, Rayner J, McHenry AM, Adams JH (2006) The structure of the Plasmodium falciparum EBA175 ligand domain and the molecular basis of host specificity. Trends Parasitol 22:143–145
Chesne-Seck ML, Pizarro JC, Vulliez-Le Normand B, Collins CR, Blackman MJ, Faber BW, Remarque EJ, Kocken CH, Thomas AW, Bentley GA (2005) Structural comparison of apical membrane antigen 1 orthologues and paralogues in apicomplexan parasites. Mol Biochem Parasitol 144:55–67
Chitnis CE, Blackman MJ (2000) Host cell invasion by malaria parasites. Parasitol Today 16:411–415
Crosnier C, Bustamante LY, Bartholdson SJ, Bei AK, Theron M, Uchikawa M, Mboup S, Ndir O, Kwiatkowski DP, Durainsingh MT, Rayner JC, Wright GJ (2011) Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature 480:534–7
Drummond PB, Peterson DS (2005) An analysis of genetic diversity within the ligand domains of the Plasmodium falciparum ebl-1 gene. Mol Biochem Parasitol 140:241–245
Duk M, Krotkiewski H, Stasyk TV, Lutsik-Kordovsky M, Syper D, Lisowska E (2000) Isolation and characterization of glycophorin from nucleated (chicken) erythrocytes. Arch Biochem Biophys 375:111–118
Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, Moch JK, Muster N, Sacci JB, Tabb DL, Witney AA, Wolters D, Wu Y, Gardner MJ, Holder AA, Sinden RE, Yates JR, Carucci DJ (2002) A proteomic view of the Plasmodium falciparum life cycle. Nature 419:520–526
Frevert U, Spath GF, Yee H (2008) Exoerythrocytic development of Plasmodium gallinaceum in the White Leghorn chicken. Int J Parasitol 38:655–672
Fu J, Saenz F, Reed MB, Balu B, Singh N, Blair PL, Cowman AF, Adams JH (2005) Targeted disruption of maebl in Plasmodium falciparum. Mol Biochem Parasitol 141:113–117
Garamszegi LZ (2006) The evolution of virulence and host specialization in malaria parasites of primates. Ecol Lett 9:933–940
Ghai M, Dutta S, Hall T, Freilich D, Ockenhouse CF (2002) Identification, expression, and functional characterization of MAEBL, a sporozoite and asexual blood stage chimeric erythrocyte-binding protein of Plasmodium falciparum. Mol Biochem Parasitol 123:35–45
Huelsenbeck JP, Ronquist F, Nielsen R, Bollback JP (2001) Bayesian inference of phylogeny and its impact on evolutionary biology. Science 294:2310–2314
Iezhova TA, Valkiunas G, Bairlein F (2005) Vertebrate host specificity of two avian malaria parasites of the subgenus Novyella: Plasmodium nucleophilum and Plasmodium vaughani. J Parasitol 91:472–474
Iyer J, Gruner AC, Renia L, Snounou G, Preiser PR (2007) Invasion of host cells by malaria parasites: a tale of two protein families. Mol Microbiol 65:231–249
Kappe SH, Noe AR, Fraser TS, Blair PL, Adams JH (1998) A family of chimeric erythrocyte binding proteins of malaria parasites. Proc Natl Acad Sci USA 95:1230–1235
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Lewis SE, Searle SM, Harris N, Gibson M, Lyer V, Richter J, Wiel C, Bayraktaroglir L, Birney E, Crosby MA, Kaminker JS, Matthews BB, Prochnik SE, Smithy CD, Tupy JL, Rubin GM, Misra S, Mungall CJ, Clamp ME (2002) Apollo: a sequence annotation editor. Genome Biol 3. RESEARCH0082
Loiseau C, Harrigan RJ, Robert A, Bowie RC, Thomassen HA, Smith TB, Sehgal RN (2012) Host and habitat specialization of avian malaria in Africa. Mol Ecol 21:431–441
Loiseau C, Iezhova T, Valkiunas G, Chasar A, Hutchinson A, Buermann W, Smith TB, Sehgal RN (2010) Spatial variation of haemosporidian parasite infection in African rainforest bird species. J Parasitol 96:21–29
Martin MJ, Rayner JC, Gagneux P, Barnwell JW, Varki A (2005) Evolution of human–chimpanzee differences in malaria susceptibility: relationship to human genetic loss of N-glycolylneuraminic acid. Proc Natl Acad Sci USA 102:12819–12824
Martinsen ES, Perkins SL, Schall JJ (2008) A three-genome phylogeny of malaria parasites (Plasmodium and closely related genera): evolution of life-history traits and host switches. Mol Phylogenet Evol 47:261–273
Mayer DC, Cofie J, Jiang L, Hartl DL, Tracy E, Kabat J, Mendoza LH, Miller LH (2009) Glycophorin B is the erythrocyte receptor of Plasmodium falciparum erythrocyte-binding ligand, EBL-1. Proc Natl Acad Sci USA 106:5348–5352
Michon P, Stevens JR, Kaneko O, Adams JH (2002) Evolutionary relationships of conserved cysteine-rich motifs in adhesive molecules of malaria parasites. Mol Biol Evol 19:1128–1142
Nagao E, Arie T, Dorward DW, Fairhurst RM, Dvorak JA (2008) The avian malaria parasite Plasmodium gallinaceum causes marked structural changes on the surface of its host erythrocyte. J Struct Biol 162:460–467
Noe AR, Adams JH (1998) Plasmodium yoelii YM MAEBL protein is coexpressed and colocalizes with rhoptry proteins. Mol Biochem Parasitol 96:27–35
Orlandi PA, Klotz FW, Haynes JD (1992) A malaria invasion receptor, the 175-kilodalton erythrocyte binding antigen of Plasmodium falciparum recognizes the terminal Neu5Ac (alpha 2–3) Gal-sequences of glycophorin A. J Cell Biol 116:901–909
Outlaw DC, Ricklefs RE (2011) Rerooting the evolutionary tree of malaria parasites. Proc Natl Acad Sci USA 108:13183–13187
Patra KP, Johnson JR, Cantin GT, Yates JR 3rd, Vinetz JM (2008) Proteomic analysis of zygote and ookinete stages of the avian malaria parasite Plasmodium gallinaceum delineates the homologous proteomes of the lethal human malaria parasite Plasmodium falciparum. Proteomics 8:2492–2499
Preiser P, Renia L, Singh N, Balu B, Jarra W, Voza T, Kaneko O, Blair P, Torii M, Landau I, Adams JH (2004) Antibodies against MAEBL ligand domains M1 and M2 inhibit sporozoite development in vitro. Infect Immun 72:3604–3608
Ricklefs RE, Outlaw DC (2010) A molecular clock for malaria parasites. Science 329:226–229
Rincon-Arano H, Guerrero G, Valdes-Quezada C, Recillas-Targa F (2009) Chicken alpha-globin switching depends on autonomous silencing of the embryonic pi globin gene by epigenetics mechanisms. J Cell Biochem 108:675–687
Saenz FE, Balu B, Smith J, Mendonca SR, Adams JH (2008) The transmembrane isoform of Plasmodium falciparum MAEBL is essential for the invasion of Anopheles salivary glands. PLoS One 3:e2287
Silva JC, Egan A, Friedman R, Munro JB, Carlton JM, Hughes AL (2011) Genome sequences reveal divergence times of malaria parasite lineages. Parasitology 138:1737–1749
Singh N, Preiser P, Renia L, Balu B, Barnwell J, Blair P, Jarra W, Voza T, Landau I, Adams JH (2004) Conservation and developmental control of alternative splicing in maebl among malaria parasites. J Mol Biol 343:589–599
Suyama M, Torrents D, Bork P (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res 34:W609–W612
Tolia NH, Enemark EJ, Sim BK, Joshua-Tor L (2005) Structural basis for the EBA-175 erythrocyte invasion pathway of the malaria parasite Plasmodium falciparum. Cell 122:183–193
Valkiūnas G, Iezhova TA, Loiseau C, Smith TB, Sehgal RN (2009) New malaria parasites of the subgenus Novyella in African rainforest birds, with remarks on their high prevalence, classification and diagnostics. Parasitol Res 104:1061–1077
Vanriper C, Vanriper SG, Goff ML, Lair M (1986) The epizootiology and ecological significance of malaria in Hawaiian land birds. Ecological Monographs 56:327–344
Verra F, Chokejindachai W, Weedall GD, Polley SD, Mwangi TW, Marsh K, Conway DJ (2006a) Contrasting signatures of selection on the Plasmodium falciparum erythrocyte binding antigen gene family. Mol Biochem Parasitol 149:182–190
Verra F, Polley SD, Thomas AW, Conway DJ (2006b) Comparative analysis of molecular variation in Plasmodium falciparum and P. reichenowi maebl gene. Parassitologia 48:567–572
Wertheimer SP, Barnwell JW (1989) Plasmodium vivax interaction with the human Duffy blood group glycoprotein: identification of a parasite receptor-like protein. Exp Parasitol 69:340–350
Acknowledgments
This work was supported by a National Institute of Health grant SC2AI089120-01A1 and the Bridge to the Doctorate grant #2R25-GM048972-11. We thank Dr. Ute Frevert and Steve Sullivan (New York University) for the P. gallinaceum samples and assistance with bioinformatics data. We also thank Shiho Kawamura for initial work on the project and Claire Loiseau for her guidance and for stimulating discussions.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 137 kb)
Rights and permissions
About this article
Cite this article
Martinez, C., Marzec, T., Smith, C.D. et al. Identification and expression of maebl, an erythrocyte-binding gene, in Plasmodium gallinaceum . Parasitol Res 112, 945–954 (2013). https://doi.org/10.1007/s00436-012-3211-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-012-3211-4