
Abstract
We developed a single step duplex real-time fluorescence resonance energy transfer (FRET) PCR merged with melting curve analysis for the fast detection and differentiation of Clonorchis sinensis and Opisthorchis viverrini eggs in human fecal samples. Two species of mitochondrial NADH dehydrogenase subunit 2 (nad2) DNA elements, the 165-bp nad2 product of C. sinensis and the 209-bp nad2 product of O. viverrini, were amplified by species-specific primers, and the fluorescence melting curve analyses were generated from hybrid of amplicons and two pairs of species-specific fluorophore-labeled probes. By their different fluorescence channels and melting temperatures, both C. sinensis and O. viverrini eggs in infected human fecal samples were detected and differentiated with high (100%) sensitivity and specificity. Detection limit was as little as a single C. sinensis egg and two O. viverrini eggs in 100 mg of fecal sample. The assay could distinguish the DNA of both parasites from the DNA of negative fecal samples and fecal samples with other parasitosis, as well as from the well-defined genomic DNA of human leukocytes and other parasites. It can reduce labor time of microscopic examination and is not prone to carry over contamination of agarose electrophoresis. Our duplex real-time FRET PCR method would be useful to determine the accurate range of endemic areas and/or to discover the co-endemic areas of two liver flukes, C. sinensis and O. viverrini, in Asia. This method also would be helpful for the differential diagnosis of the suspected cases of liver fluke infections among travelers who had visited the endemic countries of those parasites.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Two small liver flukes, Clonorchis sinensis and Opisthorchis viverrini, are both important pathogens of food-borne parasitic zoonoses causing serious public health issue in several endemic countries in Asia (Sripa et al. 2010). Life cycles of C. sinensis and O. viverrini are similar (Sripa et al. 2010; Hong and Fang 2012) involving two intermediate hosts (snails and cyprinoid fishes). Humans and other mammalian definitive hosts are infected by eating raw or semi-cooked cyprinoid fish; adult worms mature in the bile ducts of definitive hosts, and adults produce eggs which pass through the bile ducts and exit through the feces. Chronic infections with these liver flukes closely associate with the development of the bile duct (cholangiocarcinoma) and the liver (hepatocarcinoma) cancers in humans (Fried et al. 2011). While C. sinensis is endemic in People’s Republic of China, Korea, Taiwan and north Vietnam (Keiser and Utzinger 2009; Dorny et al. 2009; Hong and Fang 2012), O. viverrini is widespread over the Greater Mekong Basin including Lao PDR, Cambodia, central Vietnam and Thailand (Sripa et al. 2010; Keiser and Utzinger 2009). For clonorchiasis, more than 200 million people are at risk, 15–20 million are infected and 1.5–2 million present symptoms and complications (Hong and Fang 2012). For opisthorchiasis, approximately 9 million people are infected (World Health Organization 1995; Yossepowitch et al. 2004), and approximately 67.3 million people are at risk of infection (Keiser and Utzinger 2007; Sripa et al. 2010). In addition, the numbers of reported cases of clonorchiasis and opisthorchiasis in the USA have been increasing along with the influx of Asian immigrants (Fried and Abruzzi 2010) and importation of undercooked cyprinoid fishes (Stauffer et al. 2004).
Since the endemic areas of C. sinensis and O. viverrini are closely adjacent to each other (Keiser and Utzinger 2009; Sripa et al. 2010) with the discovery of co-endemic areas in Thailand (Traub et al. 2009), and since the number of travelers visiting endemic countries of those parasites has been increasing, an accurate differential diagnostic method for human clonorchiasis and opisthorchiasis should be developed (Le et al. 2006). At the moment, golden standard diagnosis of human liver fluke infections is to detect parasite eggs in feces, bile or duodenal fluid by microscopic methods. However, microscopic examination requires well-trained skillful personnel, and, even for the expert diagnosticians, sometimes it is difficult to differentiate C. sinensis and O. viverrini eggs as well as to discriminate opisthorchiid eggs from lecithodendriid and heterophyid parasites eggs because of their morphological similarities. To overcome the pitfalls of the microscopic methods, various sensitive and specific molecular methods have been developed to discriminate C. sinensis and O viverrini eggs. Some of the examples are multiplex PCR approaches (Le et al. 2006), PCR and sequencing of nuclear ribosomal and mitochondrial DNA (Park 2007) and nuclear marker sequences (PM-int9) (Shekhovtsov et al. 2009), PCR-RFLP analysis of the 18S-ITS1-5.8S nuclear ribosomal DNA region (Kang et al. 2008), and PCR targeting ribosomal DNA ITS regions (Sato et al. 2009). A rapid, high-throughput, specific, and sensitive real-time PCR has been applied for the detection of C. sinensis (Kim et al. 2009; Cai et al. 2012; Rahman et al. 2011) and O. viverrini (Intapan et al. 2008a, 2008b, 2009a; Suksumek et al. 2008; Sri-Aroon et al. 2011; Janwan et al. 2011) DNA. However, those methods have been applied for detecting both parasites in separate assays and are useful in endemic areas of either C. sinensis or O. viverrini alone but may be problematic where C. sinensis and O. viverrini are co-endemic. Recently, multiplex PCR amplification technique using multiplex ligation-dependent probe identification has been developed for discrimination of C. sinensis and O. viverrini in a single tube assay (Sun et al. 2011), but its applicability for fecal samples remains unknown. In the present study, duplex real-time fluorescence resonance energy transfer (FRET) PCR followed by melting curve analysis was developed first time for the detection/identification of C. sinensis and O. viverrini in a single assay. The performance of the method was evaluated with other defined genomic DNA controls and stool samples from healthy and other parasitosis patients. The results show that the method is accurate, sensitive and specific, and could be used for detection and differentiation of two parasites either presented in single or mixed infections.
Materials and methods
Parasite and DNA materials
C. sinensis adults were obtained from infected cats from the Thai Binh Province, Vietnam, whereas C. sinensis-infected human stool specimens (n = 8) were the portion of leftover specimens received from clonorchiasis patients in Nghia Hong-Nghia Hung, Nam Dinh Province, Vietnam, and kept in the Department of Parasitology, National Institute of Malariology, Parasitology and Entomology, Vietnam. O. viverrini adults (Khon Kaen strain, Thailand) were obtained from experimentally infected hamsters. Human stool specimens infected with O. viverrini (n = 30), hookworm (n = 5), Ascaris lumbricoides (n = 5), Trichuris trichiura (n = 5), Giardia lamblia (n = 5), Isospora spp. (n = 1), Trichostrongylus spp. (n = 1), Strongyloides stercoralis (n = 5), Capillaria philippinensis (n = 5), Taenia spp. (n = 5), intestinal lecithodendriid flukes (n = 5), and Echinostoma spp. (n = 5) were collected from leftover specimens from male and female patients of various ages who visited Srinagarind Hospital, Faculty of Medicine, Khon Kaen University, Thailand. The specimens were examined for parasites by the quantitative formalin ethyl acetate concentration technique (Elkins et al. 1986). The intensity of C. sinensis eggs in clonorchiasis human feces was presented as eggs per gram (EPG) of feces, and the range was 1167–6468 EPG with the geometric mean of 2330.7 EPG, and that of O. viverrini eggs in opisthorchiasis human feces ranged from 20 to 2800 EPG with the geometric mean of 139.2 EPG. Thirty negative fecal samples were obtained as controls from apparently healthy adults whose stool examination gave no evidence of any intestinal parasitic infection and whose history did not include eating raw or semi-cooked foods. The specimens were labeled blindly and used for extraction of DNA. Well-defined genomic DNA from human leukocytes, Schistosoma mekongi, S. japonicum, Centrocestus spp., Haplorchis taichui, Fasciola gigantica, Paragonimus heterotremus, Haplorchoides spp., Stellantchasmus spp. and animal schistosomes were used as controls. These DNA samples were kept in a DNA bank at −70°C in the Department of Parasitology, Faculty of Medicine, Khon Kaen University, until used. DNA from parasite-negative human feces and human feces infected with parasites other than C. sinensis and O. viverrini were extracted and purified as mentioned in the next section as the control for the specificity analysis.
This study was approved by the Khon Kaen University Ethics Committee for Human Research (reference number HE541243).
Preparation of specimens for duplex real-time FRET PCR
DNA samples were extracted from human stool specimens of C. sinensis (n = 8) and O. viverrini (n = 30) infections. For DNA extraction, 100 mg of each human fecal sample was mixed thoroughly with 200 μL of normal saline solution (0.85% NaCl in distilled water) and centrifuged at 8000 × g for 5 min. The supernatant was then discarded, and the fecal pellet was frozen at −20°C for 30 min. The frozen pellets were homogenized with disposable polypropylene pestles (Bellco Glass INC., Vineland, NJ), and DNA was extracted using the QIAamp® DNA stool mini kit (Qiagen, Hilden, Germany). The DNA was eluted in 100 μL of distilled water of which 5 μL was used for the real-time PCR reaction.
Limit of detection
To determine the detection limit of the real-time FRET PCR, 5 μL mixture of tenfold serial dilutions of equal concentrations (4 × 108 to four copies) of C. sinensis and O. viverrini positive control plasmids in water was used. Mixtures of tenfold serial dilutions of equal concentrations (20 to 2 × 10−5 ng) of C. sinensis and O. viverrini genomic DNA in water samples were also used for the evaluation of the analytical sensitivity. To determine the detection limit for eggs in feces, 100-mg fecal aliquots of parasite egg-negative fecal samples were separately mixed with one, two, five, ten, 30, 100 and 500 each of C. sinensis and O. viverrini eggs. Each mixed stool sample was separately used for DNA extraction by using QIAamp® DNA stool mini kit extraction as described earlier. The resultant DNA samples were then used for the real-time FRET PCR detection.
To evaluate the specificity of the method, well-defined genomic DNA from various parasites other than C. sinensis and O. viverrini were used as controls.
Real-time FRET PCR assay
For amplification and quantification, the LightCycler PCR and detection system (LightCycler 2.0; Roche Applied Science, Mannheim, Germany) was used. All primers and probes were designed using the LightCycler probe design software (Roche Applied Science), and the sequences are detailed in Table 1. The primers and probes were designed to bind to the portion of mitochondrial NADH dehydrogenase subunit 2 (nad2) genome of C. sinensis (a nucleotide position range of 543–707; accession number AY264851.1) and O. viverrini (a nucleotide position range of 1963–2171; GenBank accession number EU443833). The nad2 genes have been successfully used to design primers in conventional multiplex PCR for identification and discrimination of C. sinensis and O. viverrini (Le et al. 2006) and have adequate similarities and variations between them to design short range primers and probes for real-time FLET PCR. All primers and probes were synthesized by Sigma-Proligo (Singapore) and Tib Molbiol (Germany), respectively. The schematic diagram of the hybridization analysis used in the test is shown in Fig. 1.

Schematic illustration of the PCR primers (CS-F, CS-R, OV-F and OV-R primers), anchor, and detection probes for a the NADH dehydrogenase subunit 2 DNA from C. sinensis (GenBank accession number AY264851.1) and b the NADH dehydrogenase subunit 2 DNA from O. viverrini (GenBank accession number EU443833). The probes CSFL530 and OVFL530 were labeled with 530 fluorescein at the 3′ end and served as anchor probes for the sensor CSLC640 and OVLC705 probes, respectively. The sensor CSLC640 and OVLC705 probes were labeled with LightCycler Red 640 fluorophore (LC red 640) and LightCycler Red 705 fluorophore (LC red 705) at the 5′ end, respectively. Circle, fluorescein; double circle, LC red 640 or LC red 705
The 20-μL reaction mixture contained 1× LightCycler FastStart DNA Master HybProbe (Roche Applied Science), 2 mM MgCl2, 0.4 μM CS-F primer, 0.4 μM CS-R primer, 0.2 μM CSLC640 probe and 0.2 μM CSFL530 probe, as well as 0.4 μM OV-F primer, 0.4 μM OV-R primer, 0.2 μM OVLC705 probe, and 0.2 μM OVFL530 probe and 5 μL of DNA sample. The reaction was run by 45 cycles of denaturation at 95°C for 10 s, annealing at 56°C for 30 s and extension at 72°C for 10 s with the temperature transition rate of 20°C/s. The cycle number (Cn) was given as the number of PCR cycles needed for the fluorescence signal of the amplicons to exceed the detection threshold value. After amplification, a melting curve analysis was done by heating the product at 20°C/s to 95°C, cooling it to 62°C, keeping it at 62°C for 30 s, heating it slowly at 0.1°C/s to 80°C and cooling it at 40°C for 30 s. The color compensation for dual color analyses with LC Red 640 and LC Red 705 was done according to Technical Note No. LC19/2004 of Roche Applied Science. Each run contained at least one negative control consisting of 5 μL of distilled water. The specific amplified products were validated by conventional agarose gel electrophoresis.
C. sinensis and O. viverrini positive control plasmids
A positive DNA control plasmid of C. sinensis was constructed by cloning a PCR product of the nad2 sequence of the C. sinensis genome (GenBank accession number AY264851.1) into the pGEM-T vector (Promega, Madison, WI), according to the manufacturer’s instructions. The PCR products were obtained by conventional PCR using the primers CS-F and CS-R. A positive DNA control plasmid of O. viverrini was constructed also by the same manner using the nad2 DNA sequence of O. viverrini genome (GenBank accession number EU443833) and the primers OV-F and OV-R. The plasmids were propagated in Escherichia coli. Both nucleotide sequences of each inserted genes were sequenced in both directions; the resulting sequences showed a homologous structure to the gene sequences from which the primers were designed.
Data analysis
The diagnostic values were calculated and expressed using the standard method (Galen 1980). The correlation between the worm loads and the required Cn was analyzed by the Mcnemar’s χ 2 test.
Results
Standardization of the duplex real-time FRET PCR
The analytical sensitivity of duplex real-time FRET PCR was determined using 5 μL of tenfold serial dilutions (4 × 108—four copies) of the mixture of C. sinensis and O. viverrini positive control plasmids in distilled water. The detection limit of C. sinensis and O. viverrini was equal to four copies of each positive control plasmid (Fig. 2) which is equivalent to 2 × 10−4 ng each genomic DNA of C. sinensis and O. viverrini (“Online Supplementary Resource 1”) when considering 40 cycles as the cutoff detection limit. In terms of the detection limit of flukes eggs, as little as a single C. sinensis egg and two O. viverrini eggs mixed artificially in 100 mg of uninfected human feces could be clearly detected (“Online Supplementary Resource 2”).

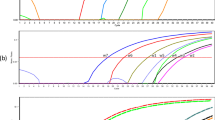
Amplification plot of fluorescence (y-axis) vs. cycle number (x-axis) showing the analytical sensitivity of real-time PCR for detecting C. sinensis (L) and O. viverrini (R) plasmid DNA. A–I Mixed tenfold dilutions of equal concentrations of C. sinensis and O. viverrini plasmids, from 4 × 108 to four copies per reaction, respectively; J distilled water (negative control)
No fluorescence signal was detected for the defined DNA controls other than C. sinensis and O. viverrini as well as with fecal samples from non-infected person or other parasitosis patients (see “Materials and methods”).
Duplex real-time FRET PCR for the detection of C. sinensis and O. viverrini in human fecal samples
The real-time FRET PCR merged with melting curve analysis yielded positive results for all the eight C. sinensis-infected and 30 O. viverrini-infected human fecal samples (for C. sinensis detection, Cn range 32.49–37.40; mean ± SD = 34.26 ± 2.10; median = 33.08 and for O. viverrini detection, Cn range 30.14–39.18; mean ± SD = 35.86 ± 1.95; median = 35.81). The mean ± SD, range and the median of the Tm values of the C. sinensis-infected human feces were 69.88 ± 0.37, 69.47–70.53 and 69.75, respectively (Fig. 3 (R)), and those of the O. viverrini-infected human fecal samples were 66.95 ± 0.54, 65.51–67.88 and 66.95, respectively (Fig. 3 (S)). However, no significant correlation was observed between the cycle numbers and the intensity of C. sinensis and O. viverrini eggs in fecal samples (P > 0.05) (data not shown). All of the control DNA samples were negative.

Melting curve analysis of two fluorophore-labeled probes hybridized to the amplification products of each NADH dehydrogenase subunit 2 (nad2) DNA from C. sinensis (P, R) and O. viverrini (Q, S). The melting temperature (Tm) of the double-stranded fragment is visualized by plotting the negative derivative of the change in fluorescence divided by the change in temperature in relation to the temperature [−(d/dT) Fluorescence (640/Back 530)] for C. sinensis detection and [−(d/dT) Fluorescence (705/Back 530)] for O. viverrini detection. The turning point of this converted melting curve results in a peak and permits easy identification of the fragment specific Tm. The results of (A) mixed C. sinensis (107 copies per reaction) and O. viverrini (107 copies per reaction) positive control plasmids, (B) non-infected human fecal samples spiked with 500 C. sinensis eggs and 500 O. viverrini eggs, (C, D) negative control containing no DNA, other parasites and other parasitosis human fecal samples (see “Materials and methods”), (E–I) clonorchiasis and (J–N) opisthorchiasis human fecal samples
Under the conditions described here, the duplex real-time FRET PCR successfully amplified a predicted 165-bp product from the DNA of the C. sinensis-infected human fecal samples (Fig. 4, lanes 1–2) and a 209-bp product from the DNA of the O. viverrini-infected human fecal samples (Fig. 4, lanes 3–4). Although some genomic DNA from other parasites and parasitosis fecal samples gave various amplified bands (“Online Supplementary Resource 3”), specific fluorescence signal was not detected by the melting curve analysis (Fig. 3).

Ethidium bromide stain patterns of the PCR products on 1.5% agarose gel. Arrows indicate the estimated 165-bp and 209-bp amplified bands of C. sinensis and O. viverrini, respectively. Lane M, DNA size markers (1 kb plus DNA ladder; Invitrogen, Carlsbad, CA); lane N, negative control containing no DNA; lane P, the PCR products obtained from mixed C. sinensis (107 copies per reaction) and O. viverrini (107 copies per reaction) positive control plasmids. Lanes 1–2, the PCR products from C. sinensis eggs-positive fecal specimens; lanes 3–4, the PCR products from O. viverrini egg-positive fecal specimens, respectively
To ensure the accuracy of the method, the amplified products from all of eight C. sinensis-infected and 30 O. viverrini-infected human fecal samples were sequenced in both directions. The results showed that all sequences were completely identical with the corresponding gene sequences from which each primer pair was designed (data not shown).
The diagnostic sensitivity and specificity of the method were all 100% for diagnosis of human clonorchaisis and opisthorchiasis.
Discussion
The real-time PCR has progressively superseded the conventional-PCR in diagnosis for infectious diseases because of its greatly improved efficacy of molecular detection. The method is not only accurate, rapid and can measure the specific DNA quantity in samples but also allows to differentiate species or strains of several pathogenic agents by melting curve analysis (Lyon and Wittwer 2009). Furthermore, this method offers a high throughput and is done in a closed system, thus eliminating the potential risk of cross-over contamination, which would occur in agarose gel electrophoresis. It can be applied as a diagnostic method and quantitative technique in parasitology (Zarlenga and Higgins 2001; Caron et al. 2008; Montes et al. 2010). The multiplex real-time PCR technique has been successfully developed for simultaneous detection and differentiation of more than one parasite in a single reaction for various parasites, i.e. intestinal protozoa (ten Hove et al. 2007, 2009; Taniuchi et al. 2011; van Lieshout and Verweij 2010), Plasmodium spp. (Khairnar et al. 2009; Veron et al. 2009; Dormond et al. 2011; Hwang et al. 2011), Wuchereria bancrofti and Brugia malayi (Intapan et al. 2009b), S. mansoni and S. haematobium (ten Hove et al. 2008), Ancylostoma duodenale, Necator americanus and Oesophagostomum bifurcum (Verweij et al. 2007), and O. viverrini and S. stercoralis (Janwan et al. 2011).
In the present study, we established a duplex real-time FRET PCR that targets mitochondrial nad2 locus of C. sinensis and O. viverrini DNA sequences, merged with melting curve analysis, for detection and differentiation of two important liver flukes in fecal samples of infected humans, in a single step assay. Two pairs of primers were used to produce species-specific amplicons, which subsequently presented different melting peak profiles generated with two pairs of different fluorophore-labeled hybridization probes. Both parasites can be differentiated by different fluorescence channels and melting temperatures. The procedure gave high sensitivity, specificity and accuracy when evaluated with a range of well-defined DNA controls as well as other control parasitosis stool samples. The real-time FRET PCR was sensitive enough to detect four copies of each artificial positive template controls. The detection limit for C. sinensis and O. viverrini genomic DNA was both 0.2 pg, which is quite similar to the detection ranges of 0.1–1 pg of C. sinensis genomic DNA (Cai et al. 2012; Rahman et al. 2011) and far smaller than that of O. viverrini genomic DNA in a real-time PCR method, which was 30 pg (Intapan et al. 2009a). The detection limit of our method was as little as one C. sinensis egg and two O. viverrini eggs in 100 mg of fecal sample, equivalent to 10 and 20 EPG, respectively. This result is similar with the egg detection limits in fecal samples of previous reports of 5–10 EPG by singleplex Taqman probe-based real-time PCR assay for C. sinensis eggs (Cai et al. 2012; Rahman et al. 2011) or of 10 EPG by singleplex FRET-probe-based real-time PCR assay for O. viverrini eggs (Intapan et al. 2009a). Variations in the detection limits noted in various reports were possibly due to the analytical sensitivity examined the parasite DNA spiked in distilled water or fecal samples. Another reason might be the use of different cutoff of cycle threshold of the method in each experiment.
In the present study, no cross-reaction was observed with the defined DNA controls other than C. sinensis and O. viverrini as well as with fecal samples from other parasitoses including lecithodendriid and heterophyid parasites, of which eggs were hard to differentiate from liver fluke eggs by microscopy. The duplex real-time FRET PCR with melting curve analysis described here would overcome the requirement of well-trained staffs for the laborious microscopic examination, because this method is not affected by the false bias inherent in microscopic examinations and eliminates any confusion with eggs of other digenean parasites. Also, many samples can be examined at the same time by this method. This in-house method is useful as the alternative tool of studying the epidemiology of clonorchiasis and opisthorchiasis as well as for eradication programs of both liver flukes in potential co-endemic areas of both parasite species. In addition, it can be used for detection/differentiation of cercariae collected in natural water reservoirs and rediae/sporocysts in infected snails, or metacercariae in cyprinoid fishes. Despite the superiorities of the methods described here, this method still requires an initial investment for the expensive equipment. This kind of costs can be covered by co-payments and compensates with other tests that use the similar technology.
In conclusion, a duplex real-time FRET PCR with melting curve analysis would be a powerful diagnostic tool for differential diagnosis of clonorchiasis and opisthorchiasis in co-endemic areas and also for the travelers wandering around the endemic areas of two parasite species.
References
Cai XQ, Yu HQ, Bai JS, Tang JD, Hu XC, Chen DH, Zhang RL, Chen MX, Ai L, Zhu XQ (2012) Development of a TaqMan based real-time PCR assay for detection of Clonorchis sinensis DNA in human stool samples and fishes. Parasitol Int 61:183–186
Caron Y, Rondelaud D, Losson B (2008) The detection and quantification of a digenean infection in the snail host with special emphasis on Fasciola sp. Parasitol Res 103:735–744
Dormond L, Jaton-Ogay K, de Vallière S, Genton B, Bille J, Greub G (2011) Multiplex real-time PCR for the diagnosis of malaria: correlation with microscopy. Clin Microbiol Infect 17:469–475
Dorny P, Praet N, Deckers N, Gabriel S (2009) Emerging food-borne parasites. Vet Parasitol 163:196–206
Elkins DB, Haswell-Elkins M, Anderson RM (1986) The epidemiology and control of intestinal helminths in the Pulicat Lake region of Southern India. I. Study design and pre- and post-treatment observations on Ascaris lumbricoides infection. Trans R Soc Trop Med Hyg 80:774–792
Fried B, Abruzzi A (2010) Food-borne trematode infections of humans in the United States of America. Parasitol Res 106:1263–1280
Fried B, Reddy A, Mayer D (2011) Helminths in human carcinogenesis. Cancer Lett 305:239–249
Galen RS (1980) Predictive value and efficiency of laboratory testing. Pediatr Clin North Am 27:861–869
Hong ST, Fang Y (2012) Clonorchis sinensis and clonorchiasis, an update. Parasitol Int 61:17–24
Hwang SY, Kim SH, Lee GY, Hang VT, Moon CS, Shin JH, Koo WL, Kim SY, Park HJ, Park HO, Kho WG (2011) A novel real-time PCR assay for the detection of Plasmodium falciparum and Plasmodium vivax malaria in low parasitized individuals. Acta Trop 120:40–45
Intapan PM, Thanchomnang T, Lulitanond V, Pongsaskulchoti P, Maleewong W (2008a) Detection of Opisthorchis viverrini in infected bithynid snails by real-time fluorescence resonance energy transfer PCR-based method and melting curve analysis. Parasitol Res 103:649–655
Intapan PM, Thanchomnang T, Lulitanond V, Phongsaskulchoti P, Maleewong W (2008b) Real-time fluorescence resonance energy transfer PCR with melting curve analysis for the detection of Opisthorchis viverrini in fish intermediate hosts. Vet Parasitol 157:65–71
Intapan PM, Thanchomnang T, Lulitanond V, Pongsaskulchoti P, Maleewong W (2009a) Rapid molecular detection of Opisthorchis viverrini in human fecal samples by real-time polymerase chain reaction. AmJTrop Med Hyg 81:917–920
Intapan PM, Thanchomnang T, Lulitanond V, Maleewong W (2009b) Rapid detection of Wuchereria bancrofti and Brugia malayi in mosquito vectors (Diptera: Culicidae) using a real-time fluorescence resonance energy transfer multiplex PCR and melting curve analysis. J Med Entomol 46:158–164
Janwan P, Intapan PM, Thanchomnang T, Lulitanond V, Anamnart W, Maleewong W (2011) Rapid detection of Opisthorchis viverrini and Strongyloides stercoralis in human fecal samples using a duplex real-time PCR and melting curve analysis. Parasitol Res 109:1593–1601
Kang S, Sultana T, Loktev VB, Wongratanacheewin S, Sohn WM, Eom KS, Park JK (2008) Molecular identification and phylogenetic analysis of nuclear rDNA sequences among three opisthorchid liver fluke species (Opisthorchiidae: Trematoda). Parasitol Int 57:191–197
Keiser J, Utzinger J (2007) Food-borne trematodiasis: current chemotherapy and advances with artemisinins and synthetic trioxolanes. Trends Parasitol 23:555–562
Keiser J, Utzinger J (2009) Food-borne trematodiases. Clin Microbiol Rev 22:466–483
Khairnar K, Martin D, Lau R, Ralevski F, Pilla IDR (2009) Multiplex real-time quantitative PCR, microscopy and rapid diagnostic immuno-chromatographic tests for the detection of Plasmodium spp performance, limit of detection analysis and quality assurance. Malar J 8:284
Kim EM, Verweij JJ, Jalili A, van Lieshout L, Choi MH, Bae YM, Lim MK, Hong ST (2009) Detection of Clonorchis sinensis in stool samples using real-time PCR. Ann Trop Med Parasitol 103:513–518
Le TH, Van De N, Blair D, Sithithaworn P, McManus DP (2006) Clonorchis sinensis and Opisthorchis viverrini: development of a mitochondrial-based multiplex PCR for their identification and discrimination. Exp Parasitol 112:109–114
Lyon E, Wittwer CT (2009) LightCycler technology in molecular diagnostics. J Mol Diagn 11:93–101
Montes M, Sawhney C, Barros N (2010) Strongyloides stercoralis: there but not seen. Curr Opin Infect Dis 23:500–504
Park GM (2007) Genetic comparison of liver flukes, Clonorchis sinensis and Opisthorchis viverrini, based on rDNA and mtDNA gene sequences. Parasitol Res 100:351–357
Rahman SM, Bae YM, Hong ST, Choi MH (2011) Early detection and estimation of infection burden by real-time PCR in rats experimentally infected with Clonorchis sinensis. Parasitol Res 109:297–303
Sato M, Thaenkham U, Dekumyoy P, Waikagul J (2009) Discrimination of O. viverrini, C. sinensis, H. pumilio and H. taichui using nuclear DNA-based PCR targeting ribosomal DNA ITS regions. Acta Trop 109:81–83
Shekhovtsov SV, Katokhin AV, Romanov KV, Besprozvannykh VV, Fedorov KP, Yurlova NI, Serbina EA, Sithithaworn P, Kolchanov NA, Mordvinov VA (2009) A novel nuclear marker, Pm-int9, for phylogenetic studies of Opisthorchis felineus, Opisthorchis viverrini, and Clonorchis sinensis (Opisthorchiidae, Trematoda). Parasitol Res 106:293–297
Sri-Aroon P, Intapan PM, Lohachit C, Phongsasakulchoti P, Thanchomnang T, Lulitanond V, Hiscox A, Phompida S, Sananikhom P, Maleewong W, Brey PT (2011) Molecular evidence of Opisthorchis viverrini in infected bithyniid snails in the Lao People’s Democratic Republic by specific hybridization probe-based real-time fluorescence resonance energy transfer PCR method. Parasitol Res 108:973–978
Sripa B, Kaewkes S, Intapan PM, Maleewong W, Brindley PJ (2010) Food-borne trematodiases in Southeast Asia epidemiology, pathology, clinical manifestation and control. Adv Parasitol 72:305–350
Stauffer WM, Sellman JS, Walker PF (2004) Biliary liver flukes (opisthorchiasis and clonorchiasis) in immigrants in the United States: often subtle and diagnosed years after arrival. J Travel Med 11:157–159
Suksumek N, Leelawat K, Leelawat S, Russell B, Lek-Uthai U (2008) TaqMan real-time PCR assay for specific detection of Opisthorchis viverrini DNA in Thai patients with hepatocellular carcinoma and cholangiocarcinoma. Exp Parasitol 119:217–224
Sun J, Xu J, Liang P, Mao Q, Huang Y, Lv X, Deng C, Liang C, de Hoog GS, Yu X (2011) Molecular identification of Clonorchis sinensis and discrimination with other opisthorchid liver fluke species using multiple Ligation-depended Probe Amplification (MLPA). Parasit Vectors 4:98
Taniuchi M, Verweij JJ, Noor Z, Sobuz SU, Lieshout L, Petri WA Jr, Haque R, Houpt ER (2011) High throughput multiplex PCR and probe-based detection with Luminex beads for seven intestinal parasites. AmJTrop Med Hyg 84:332–337
ten Hove R, Schuurman T, Kooistra M, Möller L, van Lieshout L, Verweij JJ (2007) Detection of diarrhoea-causing protozoa in general practice patients in the Netherlands by multiplex real-time PCR. Clin Microbiol Infect 13:1001–1007
ten Hove RJ, Verweij JJ, Vereecken K, Polman K, Dieye L, van Lieshout L (2008) Multiplex real-time PCR for the detection and quantification of Schistosoma mansoni and S. haematobium infection in stool samples collected in northern Senegal. Trans R Soc Trop Med Hyg 102:179–185
ten Hove RJ, van Esbroeck M, Vervoort T, van den Ende J, van Lieshout L, Verwei JJ (2009) Molecular diagnostics of intestinal parasites in returning travellers. Eur J Clin Microbiol Infect Dis 28:1045–1053
Traub RJ, Macaranas J, Mungthin M, Leelayoova S, Cribb T, Murrell KD, Thompson RC (2009) A new PCR-based approach indicates the range of Clonorchis sinensis now extends to Central Thailand. PLoS Negl Trop Dis 3:367
van Lieshout L, Verweij JJ (2010) Newer diagnostic approaches to intestinal protozoa. Curr Opin Infect Dis 23:488–493
Veron V, Simon S, Carme B (2009) Multiplex real-time PCR detection of P. falciparum, P. vivax and P. malariae in human blood samples. Exp Parasitol 121:346–351
Verweij JJ, Brienen EA, Ziem J, Yelifari L, Polderman AM, Van Lieshout L (2007) Simultaneous detection and quantification of Ancylostoma duodenale, Necator americanus, and Oesophagostomum bifurcum in fecal samples using multiplex real-time PCR. AmJTrop Med Hyg 77:685–690
World Health Organization (1995) Control of foodborne trematode infections. Report of a WHO Study Group. World Health Organ Tech Rep Ser 849:1–157
Yossepowitch O, Gotesman T, Assous M, Marva E, Zimlichman R, Dan M (2004) Opisthorchiasis from imported raw fish. Emerg Infect Dis 10:2122–2126
Zarlenga DS, Higgins J (2001) PCR as a diagnostic and quantitative technique in veterinary parasitology. Vet Parasitol 101:215–230
Acknowledgements
This work was supported by the National Science and Technology Development Agency (Discovery Based Development Grant), the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission, and the Faculty of Medicine, Khon Kaen University, Thailand.
Conflict of interest
This study was in compliance with the Thai law. No conflict of interest is declared.
Author information
Authors and Affiliations
Corresponding author
Appendix A. Supplementary data
Below is the link to the electronic supplementary material.
Online Supplementary Resource 1
The analytical sensitivity of real-time PCR (DOC 233 kb)
Online Supplementary Resource 2
Real-time PCR for detection of C. sinensis and O. viverrini eggs artificially mixed in uninfected human feces (DOC 141 kb)
Online Supplementary Resource 3
Ethidium bromide stained patterns of the PCR products (DOC 523 kb)
Rights and permissions
About this article
Cite this article
Sanpool, O., Intapan, P.M., Thanchomnang, T. et al. Rapid detection and differentiation of Clonorchis sinensis and Opisthorchis viverrini eggs in human fecal samples using a duplex real-time fluorescence resonance energy transfer PCR and melting curve analysis. Parasitol Res 111, 89–96 (2012). https://doi.org/10.1007/s00436-011-2804-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-011-2804-7