
Abstract
A loop-mediated isothermal amplification (LAMP) assay was developed for the diagnosis of Theileria lestoquardi infection. The primers were designed based on the clone-5 sequence of T. lestoquardi. The specificity and sensitivity of the assay were established. Analysis of the specificity showed that the selected LAMP primers amplified the target sequence from T. lestoquardi DNA successfully, while no amplification was seen with DNA from Theileria annulata, Theileria ovis, Babesia ovis, Anaplasma ovis, or ovine genomic DNA. The specificity of the LAMP product was further confirmed by restriction digestion and sequencing. The sensitivity of the LAMP assay was analyzed in comparison to PCR resulting in a detection limit of 10 fg/μl of plasmid DNA containing the clone-5 sequence. The suitability for utilizing the LAMP assay in the field for the diagnosis of T. lestoquardi infection was tested on 100 field samples collected in Sudan and compared with results obtained by PCR. The relative specificity and sensitivity of the established LAMP assay was determined to be 92.1% and 87.5%, respectively, indicating that it may be regarded as an alternative molecular diagnostic tool to PCR which could be used for epidemiological surveys on T. lestoquardi infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Theileria lestoquardi is a tick-borne protozoan parasite that causes a highly pathogenic disease in sheep known as malignant ovine theileriosis (MOT). The parasite is transmitted by Hyalomma ticks (Levine 1973). The disease was first described in Egypt in exported Sudanese sheep by Mason (1915) and has in the meantime been recorded in southeastern Europe, North Africa, the Near and Middle East and former southern USSR (Dolan 1989). Control of the disease can be achieved by chemotherapy, using theilericidal drugs such as buparvaquone (El Hussein et al. 1993) as well as tick control using acaricides (Jongejan and Uilenberg 2004). In addition, immunoprophylaxis of sheep with attenuated T. lestoquardi schizont-infected ovine cells has been carried out in Iraq and Iran (Hawa et al. 1981; Hooshmand-Rad 1985; Hashemi-Fesharki 1997).
Diagnosis of T. lestoquardi infection is based on the combination of the clinical signs or pathological findings with the demonstration of parasitic stages in blood or organ smears. The provisional diagnosis includes case history, clinical signs, postmortem findings, and geographic distribution of disease and vector (OIE 2000). Generally it is difficult to discriminate T. lestoquardi piroplasms from nonpathogenic Theileria species that may occur simultaneously within the same ovine host and could confuse accurate diagnosis of T. lestoquardi. Although useful in the detection of acute cases, this diagnostic procedure has limited value in determining carrier status.
Detecting antibodies against T. lestoquardi using immunofluorescence antibody technique was applied in epidemiological surveys (Salih et al. 2003; Taha et al. 2003), but false positive or false negative results due to cross-reactions or a weak specific immune response are some disadvantages that are commonly observed in this test (Leemans et al. 1997). An enzyme-linked immunosorbent assay (ELISA) has been developed for the serological detection of T. lestoquardi using recombinant protein to minimize the chance for cross-reactivity (Bakheit et al. 2006a). This ELISA is based on the newly discovered clone 5 surface protein of T. lestoquardi and was applied in field samples collected from northern Sudan (Bakheit et al. 2006a).
Several molecular techniques for detection of ovine Theileria species have been developed. PCR was developed using specific primers to amplify the T. lestoquardi fragment of the gene coding for a 30-kDa merozoite surface protein from infected ticks, sheep, and goats (Kirvar et al. 1998). The advantage of this PCR is its ability to differentiate between T. lestoquardi and Theileria annulata in the Hyalomma vector and in sheep and goats (Leemans et al. 1999a, b); however, cross-reactivity in PCR between other ovine or caprine Theileria or Babesia species was not tested (Kirvar et al. 1998), and its sensitivity is poor to detect subclinical infections. In order to overcome these limitations, a reverse line blot assay was developed for detection of Theileria and Babesia parasites infecting small ruminants (Schnittger et al. 2004). However, this method is expensive and requires sophisticated laboratory equipment, thus it is not widely applied in developing countries where the disease is endemic.
Loop-mediated isothermal amplification of DNA (LAMP) is a novel method, whereby DNA is amplified with high specificity, efficiency, and rapidity under isothermal conditions using a set of four or six specifically designed primers, which recognize six or eight specific target sequences, and a DNA polymerase with strand displacement activity (Notomi et al. 2000; Nagamine et al. 2002). LAMP can be applied using non-denatured template, and DNA extraction may also be neglected, since a drop of blood spotted on a filter paper meets the requirements to initiate the reaction (Nagamine et al. 2001).
The LAMP method has been successfully developed for the detection of some Theileria species such as Theileria parva, T. annulata, Theileria luwenshuni, and Theileria uilenbergi (Thekisoe et al. 2010; Salih et al. 2008; Liu et al. 2008). Since it is rapid and simple to run, cost effective, sensitive, and specific, the respective development of LAMP for detection T. lestoquardi can be of potential usefulness for application in diagnostics and epidemiological studies. In this study, the LAMP method was developed for detection of T. lestoquardi infection of sheep and the sensitivity and the specificity of the test were evaluated. Furthermore, the LAMP assay was applied for the diagnosis of T. lestoquardi in field samples and compared with PCR.
Materials and methods
LAMP primer design
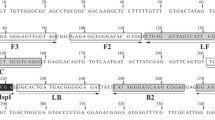
Four specific primers (F3, B3, FIP, and BIP) were derived from the sequence of the T. lestoquardi clone-5 gene (DQ004500, Bakheit et al. 2006a) using the Primer Explorer V4 program (http://primerexplorer.jp/elamp4.0.0/index.html). Since a homologue of clone-5 exists in T. annulata (Bakheit et al. 2008), the primer sets were designed for a variable region between T. lestoquardi and T. annulata. The primers were synthesized by Eurofins MWG Operon (Eurofins MWG, Ebersberg, Germany). The sequence of each primer and location within the target sequence are shown in Table 1 and Fig. 1, respectively.

Positions of the LAMP primers on the T. lestoquardi clone-5 sequence (DQ004500). The highlighted box denotes the recognition site for the Sau3AI restriction enzyme. The outer F3 and B3 primers were also used in the PCR reaction
Preparation of the DNA template
Genomic DNA was isolated from T. lestoquardi (Atbara) cell culture (Bakheit et al. 2006b) using the Gentra Puregene Cell Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. DNA from field samples was extracted using a Genomic DNA Extraction Kit (Qiagen) according to the manufacturer's instructions. The concentration of isolated DNA was assessed spectrophotometrically and diluted to 100 ng/μl, which served as template for LAMP and PCR. Genomic DNA (gDNA) of Theileria ovis, T. annulata, and Babesia ovis was obtained from the Division of Veterinary Infection Biology and Immunology of the Research Center Borstel, and gDNA of Anaplasma ovis was prepared in the Lanzhou Veterinary Research Institute.
LAMP reaction conditions and detection of the LAMP product
The reaction was performed in a final volume of 25 μl which contained 12.5 μl 2× LAMP reaction buffer [20 mM Tris–HCl (pH 8.8), 10 mM KCl, 10 mM (NH4)2SO4, 8 mM MgSO4, and 0.2% Tween 20], 125 μM each deoxynucleoside triphosphate, 0.8 M betaine (Sigma, Deisenhofen, Germany), 8 U of the Bst DNA polymerase large fragment (New England Biolabs, Frankfurt, Germany), 40 pmol each FIP and BIP primer, 5 pmol each F3 and B3 primer, and 2 μl of target DNA. The mixture was incubated at 63°C for 60 min using a conventional heating block (Stuart Scientific, Chelmsford, UK) and then heated at 80°C for 3 min to terminate the reaction. An aliquot of 5 μl of the LAMP product was subjected to electrophoresis on a 1.5% agarose gel at 90 V for 1 h and then visualized under UV light after staining with ethidium bromide.
Restriction digestion of the LAMP product
On the basis of the restriction map of the target sequence, the Sau3AI enzyme (Invitrogen, Darmstadt, Germany) was selected for use for restriction analysis of the LAMP product. Restriction with this enzyme was performed for 3 h at 37°C. The digested product was analyzed by electrophoresis on a 1.5% agarose gel as described above.
PCR verification of target length and sequencing
The outer LAMP primer pairs designated F3 and B3 were used for PCR amplification to verify whether a correct target was amplified. The reaction was performed according to Schnittger et al. (2004) with the following modifications: the primers were applied in a concentration of 10 pmol, the annealing temperature was set at 63°C, and a total number of 35 cycles were performed. The PCR product was analyzed on a 1.5% agarose gel as described above. For cloning and sequencing, the PCR DNA band was purified from agarose gel using Qiaex Gel Extraction Kit (Qiagen), cloned into the pDrive vector (Qiagen) and transformed into Escherichia coli cells. Selected clones were subjected to sequencing (Eurofins MWG Operon, Ebersberg, Germany).
Application of LAMP for field study
To evaluate the feasibility of the LAMP assay for diagnosis of field samples, a total of 100 blood samples were collected from sheep in an endemic region of northern Sudan (Atbara, n = 40) and western Sudan (Nyala, n = 60). Genomic DNA was isolated as described above. Samples were subjected to the LAMP assay and to PCR using specific primers for T. lestoquardi (Kirvar et al. 1998) for a means of comparison between LAMP and PCR results. Specificity and sensitivity were calculated as follows: specificity (Sp = number of samples negative in both tests/total number of negative samples from both tests × 100); sensitivity (Se = number of samples positive in both tests/total number of positive samples from both tests × 100).
Results
The specificity of the designed LAMP primers was examined by testing 100 ng/μl of genomic DNA of T. lestoquardi, two T. ovis isolates (from Sudan and Turkey), T. annulata, B. ovis, A. ovis, as well as sheep genomic DNA. It could be shown that the typical LAMP products appearing as a ladder-like pattern on the agarose gel were amplified only from T. lestoquardi gDNA using the designed primer set (Fig. 2a). The specificity of the LAMP product was confirmed by restriction digestion using Sau3AI enzyme (Fig. 2b) and sequencing of the 198-bp PCR product obtained using the F3 and B3 primers, which verified the clone-5 target sequence. These results demonstrated the specificity of the designed LAMP primer sets for detection of the T. lestoquardi clone-5 gene.

Specificity of the LAMP primers for the detection of the clone-5 gene of T. lestoquardi. a Detection of LAMP products using genomic DNA as indicated and analyzed in a 1.5% agarose gel. Lanes M molecular marker, 1 T. lestoquardi, 2 T. ovis (Sudan), 3 T. ovis (Turkey), 4 T. annulata, 5 B. ovis, 6 A. ovis, 7 sheep, 8 water control. b Restriction enzyme digestion of the LAMP product obtained from T. lestoquardi DNA with Sau3AI. Lanes M molecular marker, 1 unrestricted LAMP product, 2 Sau3AI restricted LAMP product
To determine the analytical sensitivity of the LAMP assay, 10-fold dilutions were made from 1 ng/μl to 1 fg/μl of plasmid DNA containing the clone-5 sequence and used as templates for the LAMP reaction (Fig. 3a). The same dilutions were also used as templates for PCR using F3 and B3 LAMP primers (Fig. 3b). The detection limit of LAMP was 10 fg/μl compared with 100 fg/μl for PCR, indicating that under the conditions used the LAMP assay has a 10-fold higher sensitivity than PCR.

Sensitivity of LAMP primers (a) and PCR primers (b) for detection of the T. lestoquardi clone-5 gene using 10-fold serial dilutions of plasmid DNA containing the clone-5 gene. a Lanes M molecular marker, 1 1 ng, 2 100 pg, 3 10 pg, 4 1 pg, 5 100 fg, 6 10 fg, 7 1 fg, 8 water control. b Lanes M molecular marker, 1 1 ng, 2 100 pg, 3 10 pg, 4 1 pg, 5 100 fg, 6 10 fg, 7 water control
A total of 100 field samples were collected from an MOT endemic region in northern and western Sudan. DNA extracted from these samples was subjected to LAMP and PCR. Table 2 presents the comparison between results obtained by PCR and LAMP. Both techniques reported similar results, where LAMP detected T. lestoquardi DNA in 27 samples out of 100 (27%) and PCR detected a total of 24 samples (24%). However, three samples were tested positive in PCR but were negative in LAMP. Likewise, there were six samples that tested positive by LAMP but were negative by PCR. The two tests agreed on 70 animals designated negative and 21 animals designated positive. This comparative evaluation revealed a relative specificity and sensitivity of the LAMP assay of 92.1% and 87.5%, respectively.
Discussion
Malignant theileriosis of sheep and goats caused by T. lestoquardi is considered among the most important tick-borne diseases of small ruminants and constitutes an obstacle to sheep industry in many developing countries like Sudan. The goal of this work was to develop a LAMP assay for the diagnosis of MOT based on the sequence of a T. lestoquardi antigenic protein clone-5 (Bakheit et al. 2006a). This protein was recently described and is known to be an immunogenic protein found on the surface of schizonts of T. lestoquardi, and the application of this clone-5 protein in ELISA for the serological diagnosis of T. lestoquardi infection was described (Bakheit et al. 2006a).
There are a number of advantages of LAMP over conventional molecular techniques. It amplifies DNA with high efficiency under isothermal conditions, it is highly specific for the target sequence, and it is simple and easy to perform once the appropriate primers are prepared (Notomi et al. 2000). Such a test could provide a useful diagnostic tool in a clinical laboratory, particularly in resources-poor countries in which malignant ovine theileriosis is a highly economically important disease. The LAMP primer set designed targeting the clone-5 gene specifically amplified T. lestoquardi target DNA, as no amplification was observed for other sheep pathogens (T. ovis, B. ovis, A. ovis) nor for the closely related pathogen T. annulata. The specificity of the clone-5 LAMP was further confirmed by restriction enzyme analysis and PCR amplification of a product of the expected size as well as sequencing of the PCR product.
The analytical sensitivity of the LAMP assay was performed using plasmid DNA containing the clone-5 target sequence. It would be of great interest to determine the sensitivity of LAMP in terms of parasitemia, which needs to be done in the future. Nevertheless, when the sensitivity of the LAMP assay was compared to that of conventional PCR, LAMP demonstrated a 10-fold higher sensitivity. Compared to PCR, LAMP does not require complicated thermal cycling steps and has the advantage of reaction simplicity and detection sensitivity. An isothermal incubation at 63°C for 1 h is enough to amplify 109 copies of the target DNA, which can easily be evaluated by visual inspection of the turbidity or fluorescence of the reaction mixture (Notomi et al. 2000; Mori et al. 2001).
To evaluate the LAMP method, 100 field samples were subjected to LAMP and PCR. In this study, 24 samples of the 100 were positive by PCR, 27 were positive for LAMP, and additionally, 6 positive samples for LAMP were negative for PCR, resulting in a sensitivity and specificity for LAMP of 87.5% and 92.1%, respectively. Therefore, this LAMP protocol gives reliable diagnostic results with a high specificity. However, further study of the LAMP method using a larger numbers of samples will help in the future to further validate the assay for application in large-scale epidemiological studies.
References
Bakheit MA, Ahmed JS, Seitzer U (2006a) A new recombinant indirect ELISA for diagnosis of malignant ovine theileriosis. Parasitol Res 98:145–149
Bakheit MA, Endl E, Ahmed JS, Seitzer U (2006b) Purification of macroschizonts of a Sudanese isolate of Theileria lestoquardi (T. lestoquardi [Atbara]). Ann N Y Acad Sci 1081:453–462
Bakheit MA, Ahmed JS, Seitzer U (2008) Existence of splicing variants in homologues of Theileria lestoquardi clone-5 genes transcripts in Theileria annulata and Theileria parva. Ann N Y Acad Sci 1149:212–213
Dolan TT (1989) Theileriosis: a comprehensive review. Rev Sci Tech Off Int Epizoot 8:11–36
El Hussein AM, El Ghali AA, Mohammed SA (1993) Efficacy of buparvaquone in the treatment of malignant theileriosis of sheep in Ed-Damer Province, N. State, Sudan. A field trial. Sud J Vet Res 12:51–57
Hashemi-Fesharki R (1997) Tick-borne diseases of sheep and goats and their related vectors in Iran. Parassitologia 39:115–117
Hawa NJ, Latif BMA, Ali SR (1981) Immunization of sheep against Theileria hirci infection with schizont propagated in tissue culture. Vet Parasitol 9:91–97
Hooshmand-Rad P (1985) The use of tissue culture attenuated live vaccine for Theileria hirci. Dev Biol Stand 62:119–127
Jongejan F, Uilenberg G (2004) The global importance of ticks. Parasitology 129:S1–S14
Kirvar E, Willie G, Katzer F, Brown CGD (1998) Theileria lestoquardi—maturation and quantification in Hyalomma anatolicum anatolicum ticks. Parasitology 117:255–263
Leemans I, Hooshmand-Rad P, Uggla A (1997) The indirect fluorescent antibody test based on schizont antigen for study of the sheep parasite Theileria lestoquardi. Vet Parasitol 69:9–18
Leemans I, Brown D, Fossum C, Hooshmand-Rad P, Kirvar E, Wilkie G, Uggla A (1999a) Infectivity and cross-immunity studies of Theileria lestoquardi and Theileria annulata in sheep and cattle: II. In vitro studies. Vet Parasitol 82:193–204
Leemans I, Brown D, Hooshmand-Rad P, Kirvar E, Uggla A (1999b) Infectivity and cross-immunity studies of Theileria lestoquardi and Theileria annulata in sheep and cattle: I. In vivo responses. Vet Parasitol 82:179–192
Levine ND (1973) Protozoan parasites of domestic animal and of man, 2nd edn. Burgess Publishing Co., Minneapolis
Liu Z, Hou J, Bakheit MA, Salih DA, Luo J, Yin H, Ahmed JS, Seitzer U (2008) Development of loop-mediated isothermal amplification (LAMP) assay for rapid diagnosis of ovine theileriosis in China. Parasitol Res 103:1407–1412
Mason FE (1915) Annual report (1914) Veterinary Service, Ministry of Agriculture. Veterinary Pathology Report. Government Press 44 Cairo
Mori Y, Nagamine K, Tomita N, Notomi T (2001) Detection of loop mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem Biophys Res Commun 289:150–154
Nagamine K, Watanabe K, Ohtsuka K, Hase T, Notomi T (2001) Loop-mediated isothermal amplification reaction using a nondenatured template. Clin Chem 47:1742–1743
Nagamine K, Hase T, Notomi T (2002) Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol Cell Probes 16:223–229
Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T (2000) Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 28:e63
OIE (2000) The Office International des epizooties: manual of standards for diagnostic tests and vaccines, 4th edn. pp 725
Salih DA, El Hussein AM, Hayat M, Taha KM (2003) Survey of Theileria lestoquardi antibodies among Sudanese sheep. Vet Parasitol 111:361–367
Salih DA, Liu Z, Bakheit MA, Ali AM, EL Hussein AM, Unger H, Viljoen G, Seitzer U, Ahmed JS (2008) Development and evaluation of a loop-mediated isothermal amplification method for diagnosis of tropical theileriosis. Transbound Emerg Dis 55:238–243
Schnittger L, Yin H, Qi B, Gubbels MJ, Beyer D, Niemann S, Jongejan F, Ahmed JS (2004) Simultaneous detection and differentiation of Theileria and Babesia parasites infecting small ruminants by reverse line blotting. Parasitol Res 92:189–196
Taha KM, EL Hussein HS, Abdalla HS, Salih DA (2003) Theileria lestoquardi infection in goats in River Nile State: comparison of serology and blood smears. Sud J Vet Sci Anim Husb 42:197–206
Thekisoe OM, Rambritch NE, Nakao R, Bazie RS, Mbati P, Namangala B, Malele I, Skilton RA, Jongejan F, Sugimoto C, Kawazu S, Inoue N (2010) Loop-mediated isothermal amplification (LAMP) assays for detection of Theileria parva infections targeting the PIM and p150 genes. Int J Parasitol 40:55–61
Acknowledgments
AMA is the recipient of a scholarship from the Sudanese Government, and DAS received an IAEA fellowship (SUD/07020). We declare that the experiments performed and presented in this report comply with the current laws of the Federal Republic of Germany.
Author information
Authors and Affiliations
Corresponding author
Additional information
Diaeldin A. Salih and Awadia M. Ali contributed equally.
Rights and permissions
About this article
Cite this article
Salih, D.A., Ali, A.M., Liu, Z. et al. Development of a loop-mediated isothermal amplification method for detection of Theileria lestoquardi . Parasitol Res 110, 533–538 (2012). https://doi.org/10.1007/s00436-011-2518-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-011-2518-x