
Abstract
Entamoeba histolytica is known to cause intestinal and extra-intestinal disease while the other Entamoeba species are not considered to be pathogenic. However, all Entamoeba spp. should be reported when identified in clinical samples. Entamoeba polecki, Entamoeba coli, and Entamoeba hartmanii can be differentiated morphologically from E. histolytica, but some of their diagnostic morphologic features overlap. E. histolytica, Entamoeba dispar, and Entamoeba moshkovskii are morphologically identical but can be differentiated using molecular tools. We developed a polymerase chain reaction (PCR) procedure followed by DNA sequencing of specific regions of 18S rRNA gene to differentiate the Entamoeba spp. commonly found in human stools. This approach was used to analyze 45 samples from cases evaluated for the presence of Entamoeba spp. by microscopy and a real-time PCR method capable of differential detection of E. histolytica and E. dispar. Our results demonstrated an agreement of approximately 98% (45/44) between the real-time PCR for E. histolytica and E. dispar and the 18S rRNA analysis described here. Five previously negative samples by microscopy revealed the presence of E. dispar, E. hartmanii, or E. coli DNA. In addition, we were able to detect E. hartmanii in a stool sample that had been previously reported as negative for Entamoeba spp. by microscopy. Further microscopic evaluation of this sample revealed the presence of E. hartmanii cysts, which went undetected during the first microscopic evaluation. This PCR followed by DNA sequencing will be useful to refine the diagnostic detection of Entamoeba spp. in stool and other clinical specimens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Six species of the genus Entamoeba, i.e., Entamoeba histolytica, Entamoeba dispar, Entamoeba moshkovskii, Entamoeba polecki, Entamoeba coli, and Entamoeba hartmanii can be found in human stools. Of those only E. histolytica is considered pathogenic causing intestinal and extra-intestinal disease (World Health Organization 1997). Nevertheless, all Entamoeba spp. should be reported in parasitological examination of stool samples. E. histolytica, E. dispar, and E. moshkovskii, are morphologically identical but can be differentiated at molecular level (Tanyuksel and Petri 2003; Fotedar et al. 2007). E. polecki, E. coli and E. hartmanii can be differentiated morphologically from E. histolytica, but some of their diagnostic morphologic features may overlap depending on the conditions of the specimen. This task may also be challenging depending on the proficiency level of the examiner, since precise morphologic identification of Entamoeba spp. to the species level requires advanced expertise (Garcia 2007). Previous studies have demonstrated that microscopic examination for diagnosis of amebiasis may not be sensitive and specific for precise identification of Entamoeba spp. in stools (González et al. 1994; Petri et al. 2000). Nevertheless, microscopic examination is considered the gold standard method for the laboratory diagnosis of Entamoeba spp. (Garcia 2007). Currently, high titers of specific serum antibodies, DNA and antigen detection can be used to support the identification of E. histolytica at the species level or to confirm the diagnosis of amebiasis (Garcia 2007; Pritt and Clark 2008). However, such approaches are not available for the differentiation of the other Entamoeba spp. DNA-based approaches are very convenient for this type of work, since these methods can be multiplexed to allow identification of multiple pathogens simultaneously (Dunbar 2006; Ali et al. 2003; Verweij et al. 2003a; Santos et al. 2007; Ben et al. 2008). Among them, several polymerase chain reaction (PCR)-based tests have been developed for the differentiation of E. histolytica, E. dispar, and more recently for E. moshkovskii in stools. However, in some of these studies false-negative results occurred when compared with microscopic examination, due to the presence of other Entamoeba species that could not be definitively identified by these molecular tests. Thus, there is still a need for alternative methods to strengthen the identification of Entamoeba spp. in human stools. In this study, we developed a PCR procedure followed by DNA sequencing analysis to differentiate all Entamoeba spp. commonly found in human stools. The PCR primers were based on a conserved portion of the 18S rRNA spanning a region with substitutions that allowed the differentiation of all Entamoeba spp. found in human stools. By using this approach, we were able to identify Entamoeba spp. that went undetected by either molecular assays or morphologic examination.
Materials and methods
Specimens
A total of 45 DNA samples, isolated from stools or cultures containing Entamoeba species were use to evaluate the PCR test. Nineteen DNA samples were from Entamoeba sp. microscopy positive stools; nine were from cultures of E. histolytica (n = 5), E. dispar (n = 3), E. moshkovskii (n = 1), which had been isolated from Brazilian patients; 16 were from clinical samples submitted to CDC for confirmatory diagnosis from state health departments. These 44 specimens were tested for the presence of E. histolytica and E. dispar by using a TaqMan PCR (Verweij et al. 2003b; Qvarnstrom et al. 2005). In addition, the specificity of this approach was tested using 11 DNA samples from other intestinal parasites: Endolimax nana (n = 1), Blastocystis hominis (n = 4), Giardia intestinalis (n = 2), Microsporidia (n = 1), Cryptosporidium parvum (n = 2), and Cryptosporidium hominis (n = 1) stools which were negative for Entamoeba spp. by microscopy and by PCR using alternative primers (Qvarnstrom et al. 2005).
Microscopic analysis
A single fresh stool sample from each individual was collected, and one part was fixed in 5% formalin, another in PVA following standard procedures, and a third aliquot of approximately 500 µl (liquid stools) or 0.2 g from the fresh material was stored at −20 C for DNA extraction. For morphologic analysis, stools were concentrated using the formalin-ethyl acetate sedimentation method and the concentrated sample was analyzed by wet mounts and as smears stained with trichrome staining procedure following protocols described elsewhere (Garcia 2007, see also www.dpd.cdc.gov/dpdx/HTML/DiagnosticProcedures).
DNA extraction
DNA from the nine cultures and the 20 stools from Brazilian patients were extracted using a method described elsewhere (Santos et al. 2007). DNA from the 16 clinical samples analyzed at CDC were extracted from 300 to 500 µl of human infected fecal suspension, using a modified version of the FastDNA method (MP Biomedicals, Solon, OH) with further purification with the QIAquick PCR purification kit (QIAGEN Inc., Valencia, CA, USA) as previously described (da Silva et al. 1999). Purified DNA was stored at 4 C until used for the molecular analysis.
PCR amplification of Entamoeba spp.
To amplify the 18S rRNA genus-specific fragment, we used the forward primer JVF (5-GTT GAT CCT GCC AGT ATT ATA TG-3) (Verweij et al. 2003a) and a reverse primer DSPR2 (5-CAC TAT TGG AGC TGG AAT TAC-3). The amplicon produced by these primers ranged from 622 to 667 bp depending on the Entamoeba species. The PCR reactions using these primers were performed in 50 µl, using AmpliTaq Gold PCR Master Mix (Applied Biosystems, [ABI], Foster City, California) and 12 pmol of each primer. Reactions were carried over in a GeneAmp PCR System 9700 thermocycler (Applied Biosytems [ABI], Foster City, CA, USA). PCR cycling parameters consisted of 5 min at 95 C and 40 cycles of 30 s at 95 C, 30 s at 57 C, and 30 s at 72 C, with a final step of 2 min at 72 C. Amplified products were visualized with ethidium bromide staining after electrophoresis on 1.5% agarose gels. Amplified PCR products produced with primers JVF/DSPR2 were purified with StrataPrep™ DNA Gel Extration Kit (Stratagene, La Jolla, CA) and cloned using pCR2.1-TOPO vector as described in the protocol from the TOP 10 Kit (Invitrogen, Carlsbad, CA, USA). Cloning was performed prior to sequencing only when needed; i.e., when there was any indication that the sample contained more than one Entamoeba spp. and when the amplification product did not allow direct sequencing. To confirm the presence of E. histolytica and E. dispar in the samples studied a TAqMan real-time PCR described elsewhere was used (Verweij et al. 2003b; Qvarnstrom et al. 2005). Reactions were performed in a volume of 25 µl with Platinum Quantative PCR Super mix-UDG with ROX reference dye (Invitrogen), 0,5 μM of primers Ehd-239F∕ Ehd-88R and 0.1 μM species-specific probes; i.e., histolytica-96 T (FAM-UCAUUGAAUGAAUUGGCCAUU-BHQ1) and dispar-96 T (HEX-UUACUUACAUAAAUUGGCCACUUUG-BHQ). PCR cycling parameters for this TAqMan assay consisted of 10 min at 95 C followed by 40 cycles of 15 s at 95 C, 30 s at 60 C, and 30 s at 72 C. Amplification, detection, and data analyses were performed with Mx3000P real time PCR thermocycler (Stratagene, La Jolla, CA, USA.
DNA sequencing analysis
Amplified PCR products generated from 18S rRNA gene of Entamoaeba spp. were purified with strataPrep PCR Purification Kit (Stratagene La Jolla, CA, USA) according to the manufacturer's instructions and sequenced in both directions using the primers JVF∕DSPR2. DNA cycle-sequencing reactions were performed using BigDye v.3.1 chemistry (Applied Biosystems, Foster City, CA, USA). Sequencing reactions were purified using Multi- systems-HV plates (Milipore Billerica, MA, USA) before loaded in the ABI Prism 3100 sequence analyzer using data collection software v. 2.0 and DNA Sequence Analysis Software v. 5.1. Sequences were assembled, edited, and aligned in DNASTAR SeqMan (DNASTAR, Inc., Madison, WI), as well as in the GeneStudio suite (GeneStudio, Inc.,Suwanee, GA). To identify Entamoeba spp. at the species level the sequence obtained was used to do BLAST searches against the GenBank database and after that aligned with sequences obtained from the GenBank.
Results
Evaluation of the JVF/DSPR2 PCR assay
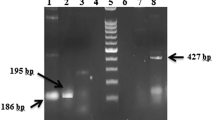
The primers used did not produce amplicons when used with DNA template from: E. nana, B. hominis, G. intestinalis, Microsporidia, C. parvum, and C. hominis. DNA samples from 45 microscopy-positive samples were used to evaluate the conventional PCR assay followed by DNA sequencing. Amplicons ranging from 622 to 667 bp, from the 18S rRNA gene of each isolate, were obtained from all these microscopy-positive samples, showing 100% sensitivity. Figure 1 shows a representation of the amplicons obtained with these primers from the different Entamoeba spp. Table 1 shows the summary of the results obtained with all the techniques employed. The agreement between the JVF/DSPR2 PCR assay and microscopy or real-time PCR for identification at species level was 89% and 98%, respectively. Eight samples were negative by the real-time PCR designed to differentiate E. histolytica from E. dispar. In these samples, we identified E. coli, E. hartmanii, or E. moshkovskii using the JVF/DSPR2 PCR, which is totally compatible with the profile of these two techniques. The results obtained from products amplified from culture samples showed 100% of agreement with the real-time PCR positive samples. The sample number 9891 was positive for E. dispar by both techniques PCR, but identification at species level was not possible through microscopic examination. This could be due to lack of cysts or trophozoites displaying enough morphologic features that could have allowed the identification at least at the complex level, i.e., E. histolytica/E. dispar.

Stained agarose gel with products amplified by PCR with primers JVF/DSPR2 from samples containing different Entamoeba. Lane 1, contains the 100-bp ladder standard. Numbers to the left of the gel are DNA fragment sizes (in base pairs). Lane 2, mix of E. histolytica/E. dispar complex with E. coli by microscopy, but E. dispar only by PCR; lanes 3 and 4, E. coli by microscopy and PCR; lanes 5, 6, and 7, E. histolytica/E. dispar complex by morphology and E. dispar by PCR; lane 8, PCR blank; lane 9, sample negative for Entamoeba sp.
Samples with more than one Entamoeba spp.
We were able to identify samples with more than one species of Entamoeba by performing sequencing analysis of the fragment amplified with primers JVF/DSPR2 (Table 1). In samples 04, 17, 19, and 3182, we identified either E. dispar/E. hartmanii (samples 17, 19, and 3192) or E. dispar/E. coli (sample 04). These results partially agreed with microscopy results, since E. hartmanii was found by microscopy in sample 3192 and E. coli was found in sample 04 in addition to E. histolytica/E. dispar complex. Nevertheless, sample 3192 was also positive for E. coli by microscopy. We believe that this was due to distortions of the morphologic features of Entamoeba found in this sample. The same explanation could be applied for samples 17 and 19. A few samples showed discrepant results between the real-time PCR and the sequencing analysis of the JVF/DSPR2 amplicon; i.e., samples 22, 26, and 204. Samples 22 and 26 were positive for E. histolytica and E. dispar by real-time PCR, but only E. dispar was identified through sequencing of the JVF/DSPR2 fragment. These results could be due to lack of sensitivity of the JVF/DSPR2 PCR for amplification of E. histolytica in these samples. Sample 204, which was positive for E. dispar, by the real-time PCR technique, had E. hartmanii based on the sequencing analysis. This result was obtained since the JVF/DSPR2 PCR could only amplify E. hartmanii. This last explanation is more compatible with the microscopic analysis, which identify E. histolytica/E. dispar complex in addition to E. hartmanii. Samples 33 and 4093 were microscopy positive for the E. histolytica/E. dispar complex and E. polecki. E. histolytica was detected by the real-time and the JVF/DSPR2 PCR, but sequence that matched E. polecki was not amplifying in both samples. The sample 9893 was positive for E. dispar by both PCR techniques. Nevertheless, this sample was positive only for E. hartmanii by microscopy. This could be due to shrunken trophozoites or cysts might be erroneously identified as E. hartmanii.
Discussion
Diagnosis of amebiasis is made by identification of Entamoeba sp. in stools or tissue using traditional microscopic analysis and is based on observation of diagnostic features. It is well known that the examination of fecal samples by light microscopy cannot in all cases differentiate all the species of Entamoeba. Some Entamoeba spp. are morphologically identical, i.e., E. histolytica, E. dispar, and E. moshkovskii. For these, microscopy can only be used to identify them at the complex/genus level. In addition, morphologic features of these species may overlap with others non-pathogenic Entamoeba, such as E. polecki, and E. hartmanii depending on the circumstances, e.g., shrunken trophozoites or cysts might be erroneously identified as E. hartmanii. Thus, alternative molecular methods could be extremely helpful to strengthen and clarify the identification of Entamoeba spp.
Many investigations have reported successful application of PCR to the diagnosis of amebiasis as a tool for confirmatory identification (Rivera et al. 1999; Mirelman et al. 1997; Evangelopoulos et al. 2000; Gonin and Trudel 2003; Kebede et al. 2004). Having methods that can be used for the accurate differential diagnostic identification of Entamoeba spp. is important for several reasons: (1) All species of Entamoeba should be reported in parasitological examination of stool samples in routine diagnosis; (2) to understand the worldwide prevalence distribution of species individually and to prevent unnecessary chemotherapy in patients with nonpathogenic species. The main objective of this investigation was to identify a region in the Entamoeba genome that could be used as a target for a PCR to amplify all species found in human stools. To address this goal, we amplified and sequenced a fragment ranging from 622 to 667 bp, corresponding to 18S rRNA gene of Entamoeba species from 45 DNA samples. The PCR assay provided discriminatory power in detecting and differentiating Entamoeba species, even when a considerable number of mixed infections were observed. However, discordant results were observed between microscopy, real time-PCR and sequencing; e.g., samples 22, 26, 3182, and 204 as described earlier. In sample 204, the most logical explanation was that it contained both E. dispar and E. hartmanii, but the molecular technique developed here was not sensitive enough to detect E. dispar. The real-time PCR Ct of this sample was 28.90 which support the hypothesis that the concentration of E. dispar DNA was enough. One possible explanation for samples 22 and 26, which were positive for both E. histolytica and E. dispar by real-time PCR and positive for E. dispar only by the JVF/DSPR2 PCR could be due to very low concentration of E histolytica DNA (The real-time PCR Ct values of 36.79 and 36.98). The sample 3182 was initially positive only for E. histolytica/E. dispar complex by microscopy. However, when this sample was re-evaluated after JVF/DSPR2 PCR results, E. hartmanii cysts were detected.
The Brazilian samples showed discrepant results between microscopy and PCR. This could be due to the fact that only one unstained sample was analyzed, which decreases the sensitivity and specificity of the morphologic analysis. In addition, E. hartmanii was not mentioned in the microscopy report because the cysts and trophozoites found were not measured. Basically, only the nuclear and cytoplasm morphology was taken in consideration for the morphologic evaluation of these samples.
Similarly results were observed by Qvarnstrom et al. (2005), which described that the real-time PCR was more sensitive than the conventional PCR techniques since the real-time PCR assay could identify E. histolytica in four clinical samples with very low parasite concentrations. However, real-time PCR assay used in this study was designed to detect only E. histolytica and E. dispar species. Interestingly, all the samples that were negative by this real-time PCR technique were positive using the JVF/DSPR2 PCR which containing Entamoeba species other than these two. In summary, we suggest that the region we identified in this study could be used to develop multiplex assays for identification of all species of Entamoeba found in human stools.
References
Ali IB, Hossain MB, Roy S, Ayeh KPF, Petri WA, Haque R, Clark CG (2003) Entamoeba moshkovskii Infections in children in Bangladesh. Emerg Infect Dis 9:580–584
Ben AS, Ben AR, Mousli M, Aoun K, Thellier M, Bouratbine A (2008) Molecular differentiation of Entamoeba histolytica and Entamoeba dispar from Tunisian food handlers with infection initially diagnosed by microscopy. Parasite 15:65–68
Da Silva AJ, Bornay LJF, Moura IN, Slemenda SB, Tuttle JL, Pieniazek NJ (1999) Fast and reliable extraction of protozoan parasite DNA from fecal specimens. Mol Diagn 4:57–64
Dunbar SA (2006) Applications of Luminex xMAP technology for rapid, high-throughput multiplexed nucleic acid detection. Clin Chim Acta 363:71–82
Evangelopoulos A, Spanakos G, Patsoula E, Vakalis N (2000) A nested multiplex PCR assay for the simultaneous detection and differentiation of Entamoeba histolytica and Entamoeba dispar in faeces. Ann Trop Med Parasitol 94:233–240
Fotedar R, Stark D, Beebe N, Mariott D, Ellis J, Harkness J (2007) Laboratory diagnosis techniques for Entamoeba species. Clin Microbiol Rev 20:511–532
Garcia LS (2007) Intestinal protozoa: amebae. In: Garcia LS (ed) Diagnostic medical parasitology, 5th edn. ASM Press, Washington DC, pp 6–25
Gonin P, Trudel L (2003) Detection and differentiation of Entamoeba histolytica and Entamoeba dispar isolates in clinical samples by PCR and enzyme-linked immunosorbent assay. J Clin Microbiol 41:237–241
González RA, Haque R, Aquirre A, Castanón G, Hall A, Guhl F, Ruiz PG, Miles MA, Warhurst DC (1994) Value of microscopy in the diagnosis of dysentery associated with invasive Entamoeba histolytica. J Clin Pathol 47:236–239
Kebede A, Verweij JJ, Petros B, Polderman AM (2004) Misleading microscopy in amoebiasis. Trop Med Int Health 9:651–652
Mirelman D, Nuchamowitz Y, Stolarsky T (1997) Comparison of use enzyme-linked immunosorbent assay-based kits and PCR amplification of rRNA genes for simultaneous detection of Entamoeba histolytica and E. dispar. J Clin Microbiol 35:2405–2407
Petri WA, Haque R, Lyerly D, Vines RR (2000) Estimating the impact of amebiasis on health. Parasitol Today 16:320–321
Pritt BS, Clark CG (2008) Amebiasis. Mayo Clin Proc 83:1154–1160
Qvarnstrom Y, James C, Xayavong M, Holloway BP, Visvesvara GS, Sriram R, Da Silva AJ (2005) Comparison of real-time PCR protocols for diferential laboratory diagnosis of amebiasis. J Clin Microbiol 43:5491–5497
Rivera WL, Tachibana H, Kanbara H (1999) Application of the Polymerase Chain Reaction (PCR) in the epidemiology of Entamoeba histolytica and Entamoeba dispar. Tokai J Exp Clin Med 23:413–415
Santos HLC, Peralta RHS, Macedo HW, Bareto MGM, Peralta JM (2007) Comparison of multiplex-PCR and antigen detection for differential diagnosis of Entamoeba histolytica. Braz J Infect Dis 11:365–370
Tanyuksel M, Petri WA (2003) Laboratory diagnosis of amebiasis. Clin Microbiol Rev 16:713–729
Verweij JJ, Laeijendecker D, Brienen EA, Van LL, Polderman AM (2003a) Detection and identification of Entamoeba species in stool samples by a reverse line hybridization assay. J Clin Microbiol 41:5041–5045
Verweij JJ, Oostvogel F, Brienen EA, Nang BA, Ziem J, Polderman AM (2003b) Prevalence of Entamoeba histolytica and Entamoeba dispar in northern ghana. Trop Med Int Health 8:1153–1156
World Health Organization (1997). Amoebiasis. Report on the WHO/Pan American Health Organization/UNESCO Expert Consultation Mexico City Geneva-WHO W Epidemiol Rec 72:97–100.
Acknowledgments
The author thanks Dr. Edward da Silva for providing cultures of E. histolytica and E. moshkovskii. Helena L C Santos was supported by a fellowship from Conselho Nacional de Desenvolvimento Cientifico e Tecnológico (CNPq) programa/Brazil. This work was also supported with funds from the National Food Safety Initiative.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Santos, H.L.C., Bandea, R., Martins, L.A.F. et al. Differential identification of Entamoeba spp. based on the analysis of 18S rRNA. Parasitol Res 106, 883–888 (2010). https://doi.org/10.1007/s00436-010-1728-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-010-1728-y