
Abstract
Glutathione (GSH) metabolism represents a potential target for antiparasitic drug design. Glutathione-S-transferase (GST), an important enzyme of the GSH cycle, is considered to be an essential detoxification enzyme in parasitic species. Soluble GST from rodent malarial parasites Plasmodium yoelii was purified to homogeneity using a combination of salt precipitation, affinity chromatography on GSH–sepharose 6B and ultrafiltration. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis revealed a single band and activity staining was also detected on PAGE gels. Kinetic studies on the purified enzyme revealed significant differences between the parasitic and mammalian enzymes. The purified enzyme exhibited an optimum pH of 8.2 and K m values of 0.2±0.213 and 3.3±0.056 mM with respect to co-substrate GSH and substrate 1-chloro-2, 4-dinitrobenzene (CDNB), respectively. Hemin, the known mammalian GST inhibitor was found to be a potent inhibitor of P. yoelii GST, with a K i of 4.0 μM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Malaria represents an emerging disease because of increasing parasite resistance against available drugs and because of increasing geographical distribution of the causative agent, Plasmodium falciparum (Deponte and Becker 2005). More than 500 million episodes of clinical malaria occur annually, with up to 2.7 million deaths (Murphy and Breman 2001), most of them in African children under the age of 5 years. Oxidative stress might play a key role in many of these fatal endpoints, and at the same time, oxidative stress represents a most promising rationale for antimalarial chemotherapy. The detoxification of reactive oxygen species (ROS) is a challenge for erythrocytes infected with Plasmodium. As a result of the high metabolic rate of the rapidly growing and multiplying parasites, large quantities of toxic redox-active by-products are generated. Apart from this metabolically derived oxidative stress, the production of ROS by the host immune system adds to the overall oxidative burden of the parasitized cell (Becker et al. 2004).
To maintain redox equilibrium, malarial parasites are equipped with a range of low molecular weight antioxidants—the most prominent being the tripeptide glutathione (GSH), as well as thioredoxin-dependent proteins (Rahlfs et al. 2002; Becker et al. 2003; Müller et al. 2003) and superoxide dismutase. Notably, Plasmodium possesses neither a classic catalase nor a classic GSH peroxidase (Sztajer et al. 2001). Some of these enzymes have been studied in functional and structural detail over the last few years and represent promising targets for the development of novel antimalarial drugs.
Glutathione-S-transferase(s) (GSTs; E.C. 2.5.1.18) are a family of phase II detoxification enzymes found in most organisms. Eukaryotes usually contain multiple GST(s) with different catalytic activities to accommodate a wide range of functions within the cells. There are two GST superfamilies: one soluble and the other membrane-bound. The soluble GST(s) occur in all aerobic organisms. Although microsomal forms have been detected (Morgenstein and DePierre 1988), an appreciable amount of GST activity has been detected in the cytosol. Cytosolic GST(s) of mammals are divided into seven classes, named alpha, mu, pi, theta, zeta, sigma, and omega, on the basis of different amino sequences and substrate specificities. Additionally, the kappa GST is a soluble mitochondrial enzyme. Studies of GST(s) from nonmammalian species have revealed the existence of several new classes, such as the beta class found in bacteria, the phi and tau-classes of plants, and the delta class described in insects (Sheehan et al. 2001). Three-dimensional structures of several members of the different GST classes were determined by X-ray crystallography and have provided insights into their unique catalytic mechanism that are not designed to accommodate any particular substrate, exhibiting a broad specificity toward substrates and nonsubstrate ligands (Listowsky 2005).
The primary function of the GST(s) lies in the protection of cellular macromolecules by catalyzing the nucleophilic addition of the thiol of reduced GSH to electrophilic centers of carcinogenic, mutagenic, toxic, and pharmacologically active compounds. GST(s) catalyze the conjugation of electrophilic xenobiotics that are subsequently metabolized to the corresponding cysteine-S-conjugates (Dekant et al. 1994), which, in turn, may be excreted unchanged or further metabolized to the mercapturic acid (Anders and Dekant 1998). Furthermore, they are also involved in metabolic pathways not associated with detoxification (Board et al. 2000; Townsend and Tew 2003; Sheehan et al. 2001; Salinas and Wong 1999). The participation of GST(s) in regulatory pathways of stress response, in the neutralization of lipid peroxidation products, in the sequestration of potentially toxic compounds, and also in drug resistance (Hemingway 2000), is especially important within a parasitic context.
GST activity has been detected in simian (Plasmodium knowlesi), rodent (Plasmodium yoelii, Plasmodium berghei), and human (P. falciparum) malarial parasites and the initial conjugation of GSH to antimalarial drugs has been described previously (Platel et al. 1999; Dubois et al. 1995; Srivastava et al. 1999). The GST of P. falciparum (Pf-GST1) has been recombinantly expressed, and substrate, as well as inhibitor profiling, has also been carried out (Harwaldt et al. 2002; Liebau et al. 2002). Unlike higher eukaryotes, Plasmodium only possesses a single gene encoding GST. Interestingly, other parasitic protozoans, such as Leishmania and Trypanosoma cruzi, also contain few GST(s) and have either unusual GSH-dependent S-transferase(s) or such S-transferase activities that depend on their unique thiol trypanothione (Ouaissi et al. 1995; Denton et al. 2004; Vickers and Fairlamb 2004). This low number of GST(s) possibly reflects that parasitic protozoans do not require a great number of xenobiotic detoxification enzymes and that GST(s) in parasitic protozoans have quite different functions from those found in other eukaryotes. The present paper reports and describes the isolation, purification, and biochemical characterization of native GST from rodent malarial parasite P. yoelii in an attempt to reveal its kinetic properties that make it different from the mammalian counterparts. The results have been discussed in terms of the utility of this enzyme as a drug target against P. falciparum in view of the critical role played by Pf-GST1 in detoxification.
Materials and methods
Chemicals
Reduced GSH, trizma, ethylene diamine tetra acetic acid (EDTA), trisodium citrate, ammonium sulfate, epoxy activated sepharose 6B, ethanolamine, sodium borate, saponin, hemin, and bovine serum albumin (BSA) were purchased from Sigma Chemical Co., St. Louis, MO, USA. All reagents used in electrophoresis and activity staining were purchased from Sigma Chemical. 1-chloro-2, 4-dinitrobenzene (CDNB) was from Spectrochem, Mumbai, India. Folin & Ciocalteu’s phenol reagent, nitro blue tetrazolium (NBT), and phenazine methosulfate (PMS) were purchased from Sisco Research, Mumbai, India. Glycerol and Giemsa stain were from Qualigens, Mumbai, India; phosphate-buffered saline (PBS) was from Himedia, Mumbai, India; and fibrous cellulose powder (CF11) was from Whatman (Kent, England). All other chemicals used were of analytical grade.
Biological materials
In vivo maintenance of rodent strain of malarial parasites P. yoelii was carried out using Swiss albino mice (Mus musculus, out-bred strain). The animals were housed in plastic cages with proper care according to the guidelines of our institutional animal house ethics committee, under standard laboratory conditions of temperature (22±1°C) and humidity (50–60%) and maintained on commercially available pellet diet supplemented with soaked grains. Water was provided ad libitum. Breeding colonies of animals were maintained under specific-pathogen-free(SPF) environment.
Plasmodium yoelii nigeriensis infection in Swiss albino mice
The laboratory isolates of P. yoelii were routinely maintained by serial blood passages in mice. Blood from infected mice was collected through cardiac puncture. The course of parasitemia was monitored daily by microscopic examination of Giemsa stained thin blood smears.
Isolation of malarial parasites
The blood of heavily P. yoelii-infected albino mice was collected in sterile citrate buffer. White cells, platelets, and plasma were removed by the passage of infected blood through a CF-11 column (Richards and Williams 1973). The malarial parasites were isolated from the erythrocytes by lysis of infected erythrocytes with 0.15% saponin and the freed parasites were collected as a pellet after centrifugation at 10,000×g for 10 min (Ferone et al. 1969). The parasites were washed several times with cold PBS (pH 7.4) and stored at −80°C till further use.
Preparation of GST from malarial parasites P. yoelii nigeriensis
For preparation of malarial GST, mixed stages of malarial parasites P. yoelii that were isolated from the blood of P. yoelii-infected Swiss albino mice in the previous step, were thoroughly washed with PBS and then homogenized with a Potter Elvehjem homogenizer in a minimum volume of PBS. The homogenate was centrifuged at 1,000×g for 15 min, 10,000×g for 30 min, and subsequently at 100,000×g for 60 min to obtain mitochondrial, post mitochondrial, and cytosolic and microsomal fractions, respectively. All the fractions obtained were analyzed for the activity of GST.
GST(s) activity determination
GST(s) activity in various dialyzed fractions was determined spectrophotometrically at 340 nm according to the method of Habig et al. (1974). All reactions were corrected for nonenzymatic conjugation, with reaction mixtures without enzyme serving as controls.
Under standard assay conditions, the reaction mixture contained 100 mM phosphate, pH 6.5, 1.0 mM CDNB in 20 μl ethanol, 1.0 mM GSH, and enzyme protein unless stated otherwise. A unit of enzyme activity was expressed as the amount that catalyzes the formation of 1 μmol S-2, 4-dinitrophenyl-GSH adduct per minute, using a molar extinction coefficient of 9.6 mM−1 cm−1 for CDNB. Protein was estimated by the method of Lowry et al. (1951) using BSA as standard.
Purification of GST
Ammonium sulfate precipitation
The cytosolic fraction of P. yoelii was subjected to ammonium sulfate fractionation (0–80%). After each fractionation, the sample was stirred in cold for 30 min and then centrifuged at 13,000×g for 15 min. All the fractions were analyzed for enrichment in GST activity. The fraction having the highest enrichment in GST activity was used as the source of GST.
GSH–sepharose affinity chromatography
Coupling of ligand to sepharose
The GSH affinity column was prepared according to the method of Simons and Vander Jagt (1977). The coupled gel was washed with 100 ml of water and the remaining active groups were blocked by allowing the gel to stand in 1.0 M ethanolamine for 4 h.
Chromatographic procedures
The protein sample was applied to the GSH–sepharose affinity column (15 ml volume), washed with 0.5 M KCl, and equilibrated with buffer A [10 mM Tris–HCl, 1.0 mM EDTA, and 3.0 mM dithiothreitol (DTT), pH 8.0] at a flow rate of 30 ml/h. After sample application, the column was washed with buffer B (10 mM Tris–HCl, 1.0 mM EDTA, 3.0 mM DTT, and 0.7 M NaCl, pH 8.0). GST was eluted from the column with buffer C (10 mM Tris–HCl, 1.0 mM EDTA, 3.0 mM DTT, and 10 mM GSH, pH 8.0).
The peak of activity determined by conjugation activity towards CDNB and absorbance at 280 nm was collected. Active fractions were pooled and concentrated about 10 times using centricon centrifugal filter devices (Millipore, Billerica, MA, USA) fitted with a 10-kDa molecular mass cut-off filter at 4,000×g for 60 min at 4°C.
Electrophoresis
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed on a 12% gel according to the method of Laemmli (1970). All reagents for SDS-PAGE were prepared and stored under specified conditions.
Activity staining
Native PAGE was performed on a 7.5% gel according to the method of Laemmli (1970). Gels were stained for GST activity after native PAGE using the method of Ricci et al. (1984). After electrophoresis, gels were placed in 0.1 M potassium phosphate buffer, pH 6.5, containing 4.5 mM GSH, 1.0 mM CDNB, and 1.0 mM NBT. Incubations were performed in a water bath thermostated at 37°C under gentle agitation. After 10 min, gels were washed with water and incubated at room temperature in 4.0 ml of 0.1 M Tris–HCI buffer, pH 9.6, containing 3.0 mM PMS. Blue insoluble formazan appeared on the gel surface in about 3–5 min, except in the GST area.
Kinetic and inhibition studies
The K m value of purified GST for substrate GSH was determined using varying GSH concentrations and a fixed CDNB concentration of 1.0 mM. The K m value for substrate CDNB was determined using varying CDNB concentrations and a fixed GSH concentration of 1.0 mM. Data were plotted as double reciprocal Lineweaver–Burk plots to determine the K m values.
To determine the dose-dependent effect of inhibitor hemin and its respective inhibitor constant (K i), enzyme was incubated with varying concentrations of inhibitor for 10 min at room temperature in the presence of 100 mM potassium phosphate buffer and 1.0 mM GSH. The reaction was initiated by the addition of 1.0 mM CDNB, and the absorbance at 340 nm was monitored for 5 min at 30-s intervals. The percentage inhibition of the enzyme activity by hemin was calculated by comparing with the enzyme activity in the absence of inhibitor.
For determining the type of inhibition, enzyme was incubated with varying concentrations of inhibitor at different substrate concentrations for 10 min at room temperature in the presence of 100 mM potassium phosphate buffer.
Results
Localization
GST activity in various subcellular fractions of P. yoelii homogenate is depicted in Table 1. GST activity in malarial parasites was found to be mainly associated with cytosolic fraction.
Purification and characterization of GST from P. yoelii
The purification scheme described here provides a convenient and highly efficient method for isolating GST from P. yoelii. Investigators have used more steps for the purification of GST, which take a long time, leading to appreciable loss in enzyme activity during the procedure. In the present investigation, therefore, a procedure was used whereby purification was carried out by the preparation of the cell lysate, salt precipitation, affinity chromatolography on GSH-sepharose, and ultrafiltration. GST purification was optimized by inclusion of relatively fewer steps to get a better yield of the enzyme. The results obtained mean that the procedure used in the purification is good enough to be used in other studies. This purification procedure also has an advantage of an experimental period as short as a day.
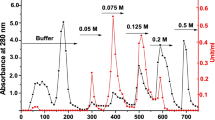
Figure 1 shows the specific activity profile of P. yoelii GST after salt fractionation. It is evident from the results that GST activity was found enriched in 55–80% ammonium sulfate fraction. Figure 2 shows the elution profile of P. yoelii GST from GSH–sepharose affinity column. Figure 3 depicts the SDS-PAGE profile, showing single-band purification of GST from P. yoelii after GSH–sepharose affinity chromatography. Figure 4 depicts activity staining of purified GST from P. yoelii.

Specific activity profile of P. yoelii GST after salt fractionation. P55: 0–55% ammonium sulfate cut precipitate, P80: 55–80% ammonium sulfate cut precipitate. Specific activity was expressed as micromoles of S-2, 4-dinitrophenyl-GSH adduct formed/min/mg±SD based on experiments done in quadruplicates

Elution profile of P. yoelii GST: GSH–sepharose affinity chromatography. E1–E12: fractions eluted by applying a gradient of 0–10 mM GSH in 10 mM Tris–HCl buffer containing 1.0 mM EDTA and 3.0 mM DTT, pH 8.0. Specific activity was expressed as micromoles of S-2, 4-dinitrophenyl-GSH adduct formed/min/mg±SD based on experiments done in quadruplicates

SDS-PAGE analysis of purified GST from P. yoelii. Lane 1: SDS marker range (14.3–97.4 kDa), Genei [lysozyme (14.3 kDa), soyabean trypsin inhibitor (20.1 kDa), carbonic anhydrase (29 kDa), ovalbumin (43 kDa), BSA (66 kDa)]. Lane 2: cytosolic fraction. Lane 3: fraction from 0–55% ammonium sulfate cut precipitate. Lane 4: fraction from 55–80% ammonium sulfate cut precipitate. Lane 5: purified GST from P. yoelii

Activity staining of purified GST from P. yoelii on 7.5% native gel
Table 2 summarizes the results of steps employed in the purification of GST from P. yoelii. GST from P. yoelii was purified to around 9.9-fold, with a total recovery of 26.9% from affinity chromatography.
Stability
The purified enzyme was fairly stable at −20°C, but there was a rapid loss of activity when the enzyme was stored at 4°C. For long-term storage, the enzyme had to be stored at −80°C without any appreciable change in its catalytic activity. Storage at 4°C for even a few days led to a complete loss of enzyme activity. The purified enzyme was highly unstable at room temperature.
Effect of pH
The optimum pH of purified P. yoelii GST was determined within the pH range of 6.0–9.5 of 0.1 M potassium phosphate buffer. Purified GST from P. yoelii showed an optimum pH centered at about 8.2 (Fig. 5).

Effect of pH of the assay buffer on purified GST from P. yoelii
Kinetic studies
The purified cytosolic GST reaction was found to be fairly linear with respect to time (30 s to 5 min), as well as the amount of enzyme protein (4.56–30.4 μg) in the case of P. yoelii (Fig. 6). Plasmodium yoelii GST displayed Michaelis–Menten behavior with regards to the co-substrate GSH and substrate CDNB. For determining K m values, the concentration of the respective substrate was systematically varied in the standard assay system at 25°C. Reciprocal plots of 1/v vs 1/[S] gave the kinetic parameters relating to one substrate in the presence of saturating concentration of the second substrate (1.0 mM). The K m values of purified P. yoelii GST with respect to GSH and CDNB were found to be 0.2±0.213 and 3.3±0.056 mM, respectively (Fig. 7).

Linearity of purified P. yoelii GST assay with respect to time and enzyme protein

Lineweaver Burk double reciprocal plot for K m of purified GST from P. yoelii with respect to GSH and CDNB
Inhibition studies
Hemin inhibited purified P. yoelii GST in a concentration-dependent fashion when studied for its effect in the concentration range 1.0–5.5 μM (Fig. 8a), showing maximum inhibition of around 86% at a 5.5-μM concentration with a K i value of around 4.0 μM (Fig. 8c). The median inhibition concentration value was 4.0 μM. Hemin exhibited noncompetitive inhibition kinetics with respect to substrate GSH (Fig. 8b).

a Inhibition profile of purified P. yoelii GST by Hemin. b Noncompetitive inhibition of Hemin on the activity of purified P. yoelii GST with respect to GSH. cKi of purified P. yoelii GST with respect to Hemin
Discussion
The present study describes the purification and characterization of GST from malarial parasites P. yoelii. The present study also attempts to delineate some of the kinetic parameters of the purified enzyme. The catalytic activity of purified GST from P. yoelii was found to be sensitive to the pH of the assay buffer. The purified enzyme showed optimal activity around pH 8.2.
The substrate specificity and inhibitor profile of P. yoelii GST appears to be different from that of GST(s) from mammalian sources. The K m value obtained with respect to GSH was 0.2 mM for P. yoelii GST, as compared to a value of 0.1–2.0 mM reported for rat liver GST(s) (Habig et al. 1974). The K m value obtained for purified P. yoelii GST with respect to GSH was quite close to that reported for P. falciparum GST, i.e., 0.164 mM (Harwaldt et al. 2002). The K m value obtained with respect to CDNB was 3.3 mM for P. yoelii GST, as compared to a value of 0.06–0.8 mM reported for rat liver GST(s) (Habig et al. 1974). The K m value obtained for purified P. yoelii GST with respect to CDNB was comparable to that reported for cloned P. falciparum GST, i.e., >2.0 mM (Harwaldt et al. 2002).
The K i of P. yoelii GST with respect to hemin (4.0 μM), the known mammalian GST inhibitor, was comparable to that of recombinant P. falciparum GST (6.5 μM). Interestingly, for hemin, a noncompetitive type of inhibition with respect to GSH was determined as compared to uncompetitive inhibition reported for recombinant P. falciparum GST (Harwaldt et al. 2002; Liebau et al. 2002). Further studies into the structure and binding properties of hemin to GST may be of critical importance for the development of therapeutic agents.
GST activity has been detected in all Plasmodium species studied so far, as well as in all intraerythrocytic stages of the parasite (Deponte and Becker 2005). The complete genome of Plasmodium was sequenced recently, revealing that the parasite harbors only one GST (Pf-GST). This observation is of particular interest: Firstly, certain antimalarial drugs such as chloroquine and methylene blue presumably influence the GSH metabolism, in which Pf-GST is involved. Secondly, Pf-GST might play a significant role in drug resistance. Pf-GST has been studied both in parasite extracts and as a recombinant protein, and its X-ray structure has been solved. The available data indicate that the homodimeric Pf-GST cannot be assigned to any of the previously known GST classes. Pf-GST exhibits significant structural differences to human GST(s), particularly at the so-called hydrophobic binding pocket (H-site) where the second substrate binds (Deponte and Becker 2005). A number of Pf-GST inhibitors including ferriprotoporphyrin IX and hemin have already been identified. The enzyme represents 1–10% of cellular protein and might therefore serve as an efficient in vivo buffer for parasitotoxic hemin, which binds uncompetitively to GST (Harwaldt et al. 2002; Liebau et al. 2002). GST of P. falciparum was found to exhibit a slight peroxidase activity that might contribute to the GSH-dependent peroxidase activity measured in malarial parasites (Becker et al. 2004). The three-dimensional structure of the enzyme has recently been elucidated at a resolution of 1.9 Å (Fritz-Wolf et al. 2003).
The three-dimensional structure of Plasmodium GST shows some similarities to Mu-class GST(s) (Fritz-Wolf et al. 2003; Perbandt et al. 2004). However, the parasite GST appears to have a larger hydrophobic binding pocket than the Mu-class mammalian GST(s), which is also more solvent-accessible because the C terminus of the protein is truncated in comparison to other GST(s). Usually, the C-terminal part of Mu-class GST(s) structurally restricts entry of substrates into the hydrophobic binding pocket, and thus, it is believed that the substrate specificity of P. falciparum GST is less restricted, which allows for the detoxification of a wider range of molecules. This can also explain why these parasites do not require as many GST(s) as do other eukaryotes. It also has been suggested that this structural difference might be exploitable for the design of parasite-specific inhibitors acting against the enzyme, and some peptide-based inhibitors have already been tested (Fritz-Wolf et al. 2003).
Pf-GST has been estimated to represent >1% of the total cellular protein (Harwaldt et al. 2002; Liebau et al. 2002). Inhibition of Pf-GST is expected to act at different vulnerable metabolic sites of the parasite in parallel. Firstly, it is likely to disturb GSH-dependent detoxification processes; secondly, it is likely to increase the levels of cytotoxic peroxides; and thirdly, it possibly increases the concentration of toxic hemin (Deponte and Becker 2005).
In the future, the significance of this protein for the survival of Plasmodium parasites can be established by genetically modifying blood stage forms. This will enhance our understanding of the detoxification processes, drug resistance, and drug action in the parasite and will open new avenues for the development of antiparasitic agents. Although the utility of this enzyme as a drug target against P. falciparum is yet to be established, the critical role played by Plasmodium GST in detoxification makes it a viable drug target against malaria.
References
Anders MW, Dekant W (1998) Glutathione-dependent bioactivation of haloalkenes. Annu Rev Pharmacol Toxicol 38:501–537
Becker K, Rahlfs S, Nickel C, Schirmer RH (2003) Glutathione: function and metabolism in the malarial parasite Plasmodium falciparum. Biol Chem 348:551–566
Becker K, Tilley L, Vennerstrom JL, Roberts D, Rogerson S, Ginsburg H (2004) Oxidative stress in malaria parasite-infected erythrocytes: host–parasite interactions. Int J Parasitol 34:163–189
Board PG, Coggan M, Chelvanayagam G, Easteal S, Jermin LS, Schulte GK, Danley DE, Hoth LR, Griffor MC, Kamath AV (2000) Identification, characterization and crystal structure of the omega class glutathione transferases. J Biol Chem 275:24798–24806
Dekant W, Vamvakas S, Anders MW (1994) Formation and fate of nephrotoxic and cytotoxic cysteine conjugates: cysteine conjugate β-lyase pathway. In: Anders MW, Dekant W (eds) Advances in pharmacology. Academic Press, London, pp 115–162
Denton H, McGregor JC, Coombs GH (2004) Reduction of antileishmanial pentavalent antimonial drugs by a parasite-specific thiol dependent reductase TDR1. Biochem J 381:405–412
Deponte M, Becker K (2005) Glutathione-S-transferase from malarial parasites: structural and functional aspects. Methods Enzymol 404:241–253
Dubois VL, Platel DF, Pauly G, Tribouley-Duret J (1995) Plasmodium berghei: implication of intracellular glutathione and its related enzyme in chloroquine resistance in vivo. Exp Parasitol 81:117–124
Ferone R, Burchall JJ, Hitchings GH (1969) Plasmodium berghei dihydrofolate reductase: isolation, properties and inhibition by antifolates. Mol Pharmacol 5:49–59
Fritz-Wolf K, Becker A, Rahlfs S, Harwaldt P, Schirmer RH, Kabsch W, Becker K (2003) X-ray structure of glutathione-S-transferase from the malarial parasite Plasmodium falciparum. Proc Natl Acad Sci USA 100:13821–13826
Habig HW, Pabst MJ, Jakoby WB (1974) Glutathione-S-transferase(s): the first enzymatic step in mercapturic acid formation. J Biol Chem 249:7130–7139
Harwaldt P, Rahlfs S, Becker K (2002) Glutathione-S-transferase of the malarial parasite Plasmodium falciparum: characterization of a potential drug target. Biol Chem 383:821–830
Hemingway J (2000) The molecular basis of two contrasting metabolic mechanisms of insecticide resistance. Insect Biochem Mol Biol 30:1009–1015
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 227:680–685
Liebau E, Bergmann B, Campbell AM, Teesdale-Spittle P, Brophy PM, Lüersen K, Walter RD (2002) The glutathione-S-transferase from Plasmodium falciparum. Mol Biochem Parasitol 124:85–90
Listowsky I (2005) A subclass of mu glutathione-S-transferases selectively expressed in testis and brain. Methods Enzymol 401:278–287
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–276
Morgenstein R, DePierre W (1988) Membrane-bound glutathione transferases. In: Sies H, Ketterer B (eds) Glutahione conjugation. Academic press, London, pp 157–175
Müller S, Liebau E, Walter RD, Krauth-Siegel RL (2003) Thiol-based redox metabolism of protozoan parasites. Trends Parasitol 19:320–328
Murphy SC, Breman JG (2001) Gaps in the childhood malaria burden in Africa: cerebral malaria, neurological sequelae, anemia, respiratory distress, hypoglycemia, and complications of pregnancy. Am J Trop Med Hyg 64:57–67
Ouaissi MA, Dubremetz JF, Schöneck R, Fernandez-Gomez R, Gomez-Corvera R, Billaut-Mulot O et al (1995) Trypanosoma cruzi: a 52 kDa protein sharing sequence homology with glutathione-S-transferase is localized in parasite organelles morphologically resembling reservosomes. Exp Parasitol 81:453–461
Perbandt M, Burmeister C, Walter RD, Betzel C, Liebau E (2004) Native and inhibited structure of a Mu class-related glutathione S-transferase from Plasmodium falciparum. J Biol Chem 279:1336–1342
Platel DF, Mangou F, Tribouley-Duret J (1999) Role of glutathione in the detoxification of ferriprotoporphyrin IX in chloroquine resistant Plasmodium berghei. Mol Biochem Parasitol 98:215–223
Rahlfs S, Schirmer RH, Becker K (2002) The thioredoxin system of Plasmodium falciparum and other parasites. Cell Mol Life Sci 59:1024–1041
Ricci G, Bello ML, Caccuri AM, Galiazzo F, Federici G (1984) Detection of glutathione-S-transferase activity on polyacrylamide gels. Anal Biochem 143:226–230
Richards WH, Williams SG (1973) The removal of leucocytes from malaria infected blood. Ann Trop Med Parasitol 67:249–250
Salinas AE, Wong MG (1999) Glutathione-S-transferase(s)—a review. Curr Med Chem 6:279–309
Sheehan D, Meade G, Foley VM, Dowd CA (2001) Structure, function and evolution of glutathione-S-transferase(s): implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J 360:1–16
Simons PC, Vander Jagt DL (1977) Purification of GST(s) from human liver by glutathione affinity chromatography. Anal Biochem 82:334–341
Srivastava P, Puri SK, Kamboj KK, Pandey VC (1999) Glutathione-S-transferase activity in malarial parasites. Trop Med Int Health 4:251–254
Sztajer H, Gamain B, Aumann KD, Slomianny C, Becker K, Brigelius-Flohé R, Flohé L (2001) The putative glutathione peroxidase gene of Plasmodium falciparum codes for a thioredoxin peroxidase. J Biol Chem 276:7397–7403
Townsend D, Tew K (2003) Cancer drugs, genetic variation and the glutathione-S-transferase gene family. Am J Pharmacogenomics 3:157–172
Vickers TJ, Fairlamb AH (2004) Trypanothione S-transferase activity in a trypanosomatid ribosomal elongation factor 1B. J Biol Chem 279:27246–27256
Acknowledgements
This investigation received financial support from Council of Scientific and Industrial Research, New Delhi (India), in the form of a Senior Research Fellowship to R. Ahmad and Volkswagen Stiftung, Hannover (Germany) in the form of ad-hoc research grant to A.K. Srivastava. We are also grateful to Dr. S.K. Puri, Head, Parasitology Division, Central Drug Research Institute, Lucknow, India, for providing the P. yoelii nigeriensis-infected albino mice for maintenance of P. yoelii infection in the present work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ahmad, R., Srivastava, A.K. Purification and biochemical characterization of cytosolic glutathione-S-transferase from malarial parasites Plasmodium yoelii . Parasitol Res 100, 581–588 (2007). https://doi.org/10.1007/s00436-006-0295-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-006-0295-8