
Abstract
The range of antioxidant enzyme systems available to Haemonchus contortus for detoxification of hydrogen peroxide was investigated using cDNA cloning of candidate genes. PCR with primers based on conserved amino acid regions and spliced leader sequences was used to obtain full-length sequences for a 2-Cys peroxiredoxin, a catalase, and a selenium-independent glutathione peroxidase, indicating that H. contortus expresses a number of antioxidant systems with the potential to detoxify peroxide (nucleotide sequence data reported in this paper are available in the GenBank, EMBL and DDBJ databases under the accession numbers AY603335, AY603336 and AY603337). Quantitative PCR analysis comparing L3-stage larvae with adult worms showed significantly elevated peroxiredoxin levels in adults, equivalent catalase levels in the two stages, and significantly less glutathione peroxidase in adults, suggesting a significant role for peroxiredoxin in allowing the nematode to detoxify hydrogen peroxide encountered in the parasitic environment. Exposure of L4-stage worms to hydrogen peroxide in vitro (generated using glucose/glucose oxidase) caused no change in mRNA levels for each of the genes, though the exposed worms showed up to eightfold higher catalase activities. The lack of mRNA changes alongside increased catalase enzyme activity indicates that transcript level was not predictive of enzyme activity, suggesting that activity may be regulated in response to oxidative stress by a mechanism other than increased transcription.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parasites encounter harmful reactive oxygen species (ROS) derived from both endogenous and exogenous sources. The ROS (including superoxide anion, hydrogen peroxide, and the hydroxy radical) are damaging to parasite lipids, proteins and nucleic acids. Normal metabolism within the parasite in an aerobic environment (for example, by the mitochondrial respiratory chain) will generate ROS as oxygen is reduced to water. As part of the host’s efforts to expel parasites, the ‘respiratory burst’ of host phagocytes (eosinophils, neutrophils and macrophages) generates ROS through the action of the enzyme NADPH oxidase (Smith 1989). In addition, it is likely that blood-feeding parasites will need to detoxify ROS derived from ingested oxy-haemoglobin.
Parasites, in common with all organisms that encounter ROS, possess a number of antioxidant enzyme systems that serve as protective mechanisms. These enzymes include superoxide dismutase (SOD), catalase, peroxiredoxin, and glutathione peroxidase (GPX) (Selkirk et al. 1998; Henkle-Duhrsen and Kampkotter 2001). SOD catalyses the conversion of superoxide anion to hydrogen peroxide, which may in turn be detoxified by peroxiredoxin, catalase or GPX. Whilst catalase activity is limited to hydrogen peroxide removal, peroxiredoxin and GPX can also catalyse the reduction of organic hydroperoxides. The mechanism of hydrogen peroxide detoxification varies amongst different parasite species; for example, catalase and GPX are absent from many parasites (Callahan et al. 1988), and peroxiredoxin is thought to fulfil the hydrogen peroxide-detoxification role in some of these species (McGonigle et al. 1998; Henkle-Duhrsen and Kampkotter 2001). In addition, cytochrome c peroxidase has been suggested as a mechanism for removal of hydrogen peroxide in a number of parasites (e.g. Campos et al. 1999).
Evidence for a role of antioxidant enzymes in parasite defence against host-generated oxidants comes largely from studies in which the susceptibility to oxidants in different parasite species, or life stages within a species, is correlated with differences in antioxidant enzyme activities in the separate species or life stages (Kazura and Meshnick 1984; Smith and Bryant 1986; Mkoji et al. 1988; Nare et al. 1990; Ou et al. 1995). More recently, several reports have demonstrated increased levels of antioxidant enzymes in response to oxidative stress (Liebau et al. 2000; Ben-Smith et al. 2002). There is some evidence that catalase plays a role in defence against hydrogen peroxide in the nematode Haemonchus contortus. Kotze and McClure (2001) showed that inhibition of H. contortus catalase (using aminotriazole) rendered the nematode more susceptible to hydrogen peroxide. A subsequent study (Kotze 2003) demonstrated that both adult and L4 H. contortus showed induced catalase activities after exposure to low levels of hydrogen peroxide, and that this increased catalase activity in adult worms was associated with a greater ability of the parasite to survive subsequent exposure to toxic levels of the peroxide (compared to controls with no pre-exposure).
The present study aimed to investigate the range of potential defences against hydrogen peroxide in H. contortus by examining free-living L3 and parasitic adult stages for the presence and relative expression levels of peroxiredoxin, catalase and GPX genes. Given that this nematode responds to oxidative stress by increasing its protective catalase activities (Kotze 2003), we also investigated whether this response was associated with increased mRNA levels for the antioxidant genes.
Materials and methods
cDNA cloning
Initial experiments to clone antioxidant genes from H. contortus were performed using RNA prepared from L3 larvae. Larvae were collected after migration from faecal cultures and washed several times in large volumes (>1 l) of water. Batches of larvae (approximately 500 mg) were washed several times in water (2 ml) and 50-mg amounts were resuspended in 0.5 ml of a guanadinium isothiocyanate/sodium citrate/sarcosyl/mercaptoethanol solution (Stratagene Micro RNA Isolation Kit), snap frozen in liquid nitrogen, and homogenized using a mortar and pestle held on dry ice. DNA and protein were removed by partition into an acidic phenol/chloroform/isoamyl alcohol solution, and the RNA was precipitated using isopropanol. Double-stranded cDNA was synthesised using the Clontech SMART PCR cDNA Synthesis Kit.
Subsequent experiments to clone peroxiredoxin, catalase and GPX genes utilized a series of PCR steps. In each case, degenerate primers were designed against conserved regions in the amino acid sequences of homologous proteins from other organisms. The products of these initial PCRs were cloned and sequenced to generate the initial sequence information for the H. contortus genes. Subsequently, these known sequences were extended using PCRs with primers from within the area of known sequence, further sets of degenerate primers, spliced leader (SL1 or SL2) primers, and a poly-T primer. The 5′ end of the cDNA was obtained using primers with the SL1 sequence (peroxiredoxin and catalase) or the SL2 sequence (GPX) as the forward primer. The 3′ end of each cDNA was obtained using a poly-T primer as the reverse primer. The PCR steps and primer sequences are shown in Table 1. These PCR steps allowed a full sequence to be constructed for each gene. In order to confirm the identity of these composites, full-length sequence was obtained by sequencing of clones for each gene derived from PCRs using primers from the 5′ and 3′ untranslated regions: peroxiredoxin, F–GGTTTAATTACCCAAGTTTGAG (=SL1), R–ATAGATTGATACACCGAGAAGAG; catalase, F–CAATTAATAAGCACGATGCC, R–CTTTGCTGACATGACAAAAC; glutathione peroxidase, F–CCAGTTACTCAAGGACCAAAG, R–GACGAAAGGAAAAGGAAAAG. These PCRs were performed using cDNA derived from adult-stage nematode RNA prepared using the RNeasy Kit (Qiagen), and the products were cloned using the TOPO TA Cloning kit (Invitrogen) (as described below).
PCRs were performed using either standard or touchdown protocols. The standard protocol consisted of an initial period of 3 min at 94°C, followed by 36 cycles of 94°C for 30 s, an annealing temperature (either 50 or 60°C) for 30 s, and 72°C for 90 s, followed by an extension for 10 min at 72°C. The touchdown protocol consisted of an initial period of 2 min at 94°C, followed by a number of cycles of 94°C for 30 s, an annealing period of 2 min at a temperature which decreased progressively by 0.5°C per cycle, and a 90-s extension at 68°C. The annealing-temperature gradient continued for either 30 cycles or 20 cycles depending on whether it extended over a 15 or a 10°C range respectively. At the conclusion of the temperature gradient, the PCR continued for a further 10 cycles (if the gradient range was 15°C) or 20 cycles (if the gradient range was 10°C) at 94°C for 30 s, at the temperature of the gradient endpoint for 60 s, and an extension at 68°C for 90 s. The final step was an extension at 68°C for 10 min. Primer concentrations were 2.5 μM for degenerate primers and 0.5 μM for specific primers. Red Hot DNA Polymerase (Advanced Biotechnologies) was used for standard PCRs, and Clontech Advantage cDNA Polymerase Mix (containing TaqStart Antibody) was used for touchdown PCRs. PCR products were cut from agarose gels and cloned using the TOPO TA Cloning kit (Invitrogen).
Life-stage patterns
L3 nematodes were collected as they migrated from sheep faeces, kept for up to 1 month at 10°C, and then returned to room temperature for an overnight period before RNA was extracted. Adult nematodes were recovered from sheep abomasa approximately 8–10 weeks after infection. The infections were maintained by weekly administration of the cortico-steroid Trimadexil (0.5 ml per sheep each week). Adult nematodes were recovered in RPMI-1640-based medium [as described by Kotze and McClure (2001)], rinsed several times in this medium, and then snap frozen in liquid nitrogen, before transfer to −70°C for storage. RNA was extracted from L3 and adults using the RNeasy kit (Qiagen) after initial homogenization using a shaped glass rod as a pestle in an Eppendorf tube. A DNase step was performed on the RNeasy column as described by the manufacturer. cDNA was synthesized using Superscript II (Life Technologies).
Exposure to oxidative stress
L4 worms were cultured in vitro as described previously (Kotze 2003). In experiment 1, the worms were cultured in rolling bottles to the L4 stage over a 5-day period, then transferred to fresh control medium or medium containing 10 mU/ml glucose oxidase in wells of a 12-well microtitre plate for a period of 48 h (worm concentration was 5,000 in 2 ml of medium). After this time, the worms were collected for enzyme activity assays and RNA determinations as described below. For experiment 2, L4 worms (after 4 days in the culture medium) were placed in fresh medium in 12-well plates and exposed to 10 mU/ml glucose oxidase (or control medium). At each subsequent 24-h time point, the worms were collected and control worms were again placed into control medium, while the GO-treated worms were placed into medium with increased GO levels at each 24-h point; that is, 20 mU/ml GO at 24–48 h, 40 mU/ml at 48–72 h, and finally 80 mU/ml GO at 72–96 h. Worms were then collected and processed for enzyme or RNA determinations.
Catalase enzyme activity
Worms were homogenized in 50 mM potassium phosphate buffer, pH 7.8, containing 0.1 mM EDTA and 1 mM PMSF. The homogenate was centrifuged at 10,000 g for 10 min, and the supernatant was used as the enzyme solution. Catalase activity was assayed as described previously (Kotze and McClure 2001) by monitoring (at 240 nm) the disappearance of hydrogen peroxide over a 30-s period. Protein was measured by the method of Bradford (1976) using bovine serum albumin (BSA) as standard.
Quantitative PCR
Quantitative PCR was performed on an ABI Prism 7900HT Sequence Detection System using the primers described in Table 2. PCRs amplifying a region of H. contortus 18S ribosomal RNA (accession number L04153) were used as a reference for calculation of relative expression levels of each of the antioxidant genes. PCR conditions were as follows: 1 cycle of 95°C for 15 min; 40 cycles of 95°C for 30 s, 62°C for 30 s, 72°C for 90 s; and for dissociation curve analysis: 1 cycle of 95°C for 15 s, 62°C for 15 s, 95°C for 15 s with a 2% ramp rate. PCRs were performed using a series of three or four ten-fold serially diluted cDNA concentrations (triplicate PCRs at each cDNA level) in order to generate standard curves for each gene. A number of standard curves (n=11–15) were performed for each gene, and the slope of each standard curve was determined (slope=Δ CT/Δ log10 cDNA level, where CT= critical threshold). The mean slope of the standard curves for each gene was calculated. ANOVA showed that significant differences (P=0.05) existed between the mean slopes for each gene, thereby necessitating the use of separate mean slope values for each gene in calculating expression levels in subsequent analyses.
For the life-stage experiment, cDNA preparations (n=3 separate preparations for each life stage) were examined in triplicate PCRs at each of three 10-fold serially diluted cDNA concentrations. The PCR analysis was performed twice (that is, 18 PCRs total for each cDNA preparation for each gene). For each GO-induction experiment, cDNA was prepared from single control or GO-treated assays. Each cDNA was examined in triplicate PCRs at each of three (experiment 1) or four (experiment 2) ten-fold serially diluted cDNA concentrations. Q-gene software (Biotechniques; http://www.biotechniques.com/softlib/qgene.html) was used to compare CT values for each gene with the CT values given by PCRs of the 18S gene performed at the same time, at equivalent cDNA concentrations. The Q-gene software generated normalized expression values for each gene with each cDNA preparation.
Significantly, preliminary PCRs showed that each of the primer pairs shown in Table 2 gave amplification of a product in PCRs using genomic DNA (prepared according to standard methods). Therefore, in order to ensure that expression-level assessments on cDNA preparations were not affected by possible contamination with genomic DNA, PCRs were performed on all cDNA preparations using primers designed from sequences within H. contortus alpha-tubulin introns 3 and 4 (forward and reverse primers respectively) (accession number X80046) (see Table 2). The PCR showed a product with genomic DNA (partial introns 3 and 4, and complete exon 4), but no product with cDNA. Absence of a product in PCRs with cDNA in the present study would, therefore, indicate an absence of significant genomic contamination in cDNA preparations.
Results and discussion
cDNA cloning experiments revealed full-length sequences for a catalase, a peroxiredoxin, and a GPX gene (submitted to GenBank with accession numbers AY603335, AY603336, AY603337 respectively). Peroxiredoxin and GPX full-length sequences were obtained after two PCR steps, whilst four steps were required to obtain the catalase sequence (Table 1).
The peroxiredoxin sequence consisted of a spliced leader (SL1) followed immediately by a start codon and an open-reading frame of 591 nt, including a stop codon. The open-reading frame was predicted to encode a protein of 196 amino acids with a calculated molecular mass of 22,000 Da. The peroxiredoxin is a member of the 2-Cys class of peroxiredoxin proteins (2-Cys PRX) with conserved cysteine residues at Cys-49 and Cys-170 (Henkle-Duhrsen and Kampkotter 2001). The gene also shows the presence of the conserved regions surrounding the two cysteine residues in 2-Cys peroxiredoxins: the FVCP sequence flanking Cys-49, and the VCP sequence flanking Cys-170. The sequence is almost identical to one of the four clusters identified for H. contortus peroxiredoxin/peroxiredoxin on the NEMBASE database (http://www.nematodes.org/) (cluster HCC00319).
The H. contortus catalase gene consisted of an SL1 sequence, followed by a start codon and an open-reading frame of 1,518 nt including a stop codon. The cDNA sequence encoded a 505-amino acid protein with a predicted molecular mass of 58,000 Da. The gene sequence shows significant similarity (at its 5′ end) to a cluster reported for a H. contortus catalase (accession number BF23075, NEMBASE cluster HCC01357; length 660 nt; sequence identity of 86% over 530 nt). The H. contortus catalase lacks the peroxisomal targeting signal (SKL or its functional variants) found at the C-terminus of most proteins that are transported to peroxisomes (Rachubinski and Subramani 1995), indicating that it is most likely located in the cytosol, and suggesting that it plays a role in defence against exogenous hydrogen peroxide. Peroxisomal localisation would be more indicative of a role in detoxification of peroxide generated in normal metabolism within the nematode. In this regard, the H. contortus catalase is similar to the cytosolic catalase in Ascaris suum, which also lacks the peroxisomal targeting signal (Eckelt et al. 1998).
The GPX peroxidase sequence consisted of an SL2 sequence, followed by a start codon and an open-reading frame of 624 nt including a stop codon. The sequence encoded a 207-amino acid protein with a predicted molecular mass of 24,100 Da. The sequence included a cysteine residue at position 50, identifying the protein as a selenium-independent GPX. In contrast, selenium-dependent GPXs show a seleno-cysteine residue encoded by the stop codon TGA at this position. The sequence was distinct from the H. contortus GPX described by Hartman et al. (2001) (NEMBASE cluster HCC01990, accession number AF305967). It is apparent that H. contortus possesses at least two distinct GPX enzymes. The previously described protein also shows a cysteine residue at the active site (position 55) identifying it as a selenium-independent enzyme. Whilst selenium-dependent GPXs would be expected to show activity with hydrogen peroxide as well as with organic hydroperoxides, the substrate specificity of selenium-independent GPXs is not as clear. Tang et al. (1995) found that a recombinant selenium-independent GPX from Brugia phangi (gp29) showed no significant activity against hydrogen peroxide. The enzyme was active towards a number of organic hydroperoxides, and was shown to suppress lipid peroxidation, suggesting that it may play a role in the repair of oxidatively damaged membranes (Tang et al. 1996) rather than acting directly as an antioxidant enzyme. In contrast, several studies have demonstrated activity of mammalian selenium-independent GPX enzymes with hydrogen peroxide (Vernet et al. 1996; Singh and Shichi 1998). In addition, a selenium-independent GPX from Dirofilaria immitis showed activity against hydrogen peroxide as well as a range of hydroperoxide substrates (Tripp et al. 1998). There is potential, therefore, for the H. contortus GPXs to be involved in detoxification of hydrogen peroxide; however, their role remains unclear in the absence of substrate specificity information.
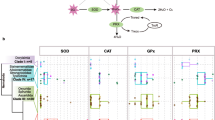
Initial quantitative PCRs showed no significant amplification of a product using primers designed for the intron of the tubulin gene (data not shown), indicating that the cDNA preparations contained no significant genomic DNA contamination. The relative expression of each of the antioxidant genes in L3 and adult stages (relative to 18S expression in each life stage) is shown in Fig. 1. Peroxiredoxin was expressed at higher levels than the other two genes in both life stages. The relative expression in adults compared to L3 was different for each gene; peroxiredoxin was increased 4.3-fold in adults compared to L3, catalase was not significantly different in the two life stages, and GPX showed a significant decrease of sevenfold in adults compared to L3. These changes in gene expression patterns would most likely indicate consequent changes in enzyme activities between the two life stages.

Expression (relative to ribosomal RNA expression levels) of peroxiredoxin, catalase and glutathione peroxidase in L3 and adult cDNA preparations from H. contortus. Data are shown as means±SE, n=3 separate RNA preparations. Each RNA was converted into a single cDNA preparation, and assayed in triplicate PCRs on two separate occasions at each of three different cDNA concentrations; that is, 18 PCRs were used to generate normalized expression results for each gene with each cDNA preparation. For statistical analyses, data were log-transformed and analysed using one-way ANOVA, and differences between means were examined using least significant difference values (P=0.05). Columns labelled with identical letters were not significantly different
The sheep from which the adult nematodes were recovered in the present study had been treated weekly with a steroid (Trimadexil) to suppress the host’s immune system in order to reduce the likelihood of parasite expulsion and guarantee the supply of nematodes as experimental samples. Whilst this approach has been useful in routinely providing adult H. contortus nematodes for study and has allowed detection of each of the antioxidant genes in the present study, it will be of more-relevant interest to examine expression patterns of the antioxidant genes in parasites exposed to the ‘normal’ host immune response, and to the response of sheep with heightened innate or acquired immunity. Therefore, with regard to the present study, the gene expression changes noted in adults compared to L3 should be viewed as indicating the relative levels of each of the antioxidant genes in the adult stage in the presence of a reduced host immune response. However, the significant increase in peroxiredoxin gene expression levels (alongside equivalent catalase and decreasing GPX) in comparing the parasitic adult to the free-living L3 suggests that peroxiredoxin may be significant in allowing the nematode to survive in the parasitic environment. The adult nematodes examined in this study would have been exposed as ‘normal’ to ROS derived from ingested oxy-haemoglobin.
Exposure of L4 larvae to hydrogen peroxide (generated using GO) caused induction of catalase activities of approximately fivefold and eightfold (on a per-mg-protein basis), in experiments 1 and 2 respectively (data not shown), similar to the induction effects reported earlier by Kotze (2003). However, this increased enzyme activity was not associated with any increase in transcript levels for each of the antioxidant genes (Fig. 2).

Expression (relative to ribosomal RNA expression levels) of peroxiredoxin, catalase and glutathione peroxidase (GPX) in cDNA preparations from L4-stage H. contortus maintained in the presence or absence of GO. Experimental conditions are described in the text. A Experiment 1. B Experiment 2. Data are shown as means±SE, n=3 (A) or 4 (B) ten-fold serially diluted cDNA concentrations (triplicate PCRs at each concentration). For statistical analyses, data were log-transformed and control or GO-treated data were compared for each gene separately using t-tests. Within each gene, columns labelled with identical letters were not significantly different (P=0.05)
While Fig. 2 shows elevated peroxiredoxin levels compared to the other two genes, as in Fig. 1, it is apparent that expression levels for each of the antioxidant genes were much lower in L4 compared to L3 and adult worms. This apparent down-regulation may be a consequence of in vitro maintenance of this parasitic worm stage, rather than an actual measure of relative gene expression across the three life stages. Hence, the actual gene expression levels shown by such experiments may not reflect the significance of particular genes in L4 stage worms in vivo under ‘normal’ parasitic environment conditions. However, despite this suggested ‘artificial’ nature of gene expression levels in L4 worms cultured in vitro, the clear ability of these larvae to respond to oxidative stress [catalase activity in this study and Kotze (2003)] suggests that this experimental system is useful for examining the relationship between gene expression and protein activity in response to stimuli.
Although changes in gene expression are often used as a measure to indicate the biological significance of particular proteins, data on the non-predictive correlation between mRNA and protein levels (Futcher et al. 1999; Gygi et al. 1999) indicate that the two measures are not linked in many cases, with increased protein activity/expression not always associated with increased transcription. In the present study, an increased catalase activity in worms following oxidative stress was not associated with any increased catalase gene expression or increased expression of the other two antioxidant genes, suggesting that the induction response to oxidative stress is not associated with increased transcription of the three genes. Antioxidant enzyme systems in different organisms are known to be regulated by a variety of mechanisms, including post-transcriptionally (e.g. Reimer et al. 1994; Clerch 2000). The mechanism of regulation of H. contortus antioxidant systems warrants further investigation.
References
Ben-Smith A, Lammas DA, Behnke JM (2002) Effect of oxygen radicals and differential expression of catalase and superoxide dismutase in adult Heligmosomoides polygyrus during primary infections in mice with differing response phenotypes. Parasite Immunol 24:119–129
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72:248–254
Callahan HL, Crouch RK, James ER (1988) Helminth anti-oxidant enzymes: a protective mechanism against host oxidants? Parasitol Today 4:218–225
Campos EG, Hermes-Lima M, Smith JM, Prichard RK (1999) Characterisation of Fasciola hepatica cytochrome c peroxidase as an enzyme with potential antioxidant activity in vitro. Int J Parasitol 29:655–662
Clerch LB (2000) Post-transcriptional regulation of lung antioxidant enzyme gene expression. Ann N Y Acad Sci 899:103–111
Eckelt VHO, Liebau E, Walter RD, Henkle-Duhrsen K (1998) Primary sequence and activity analyses of a catalase from Ascaris suum. Mol Biochem Parasitol 95:203–214
Futcher B, Latter GI, Monardo P, McLaughlin CS, Garrels JI (1999) A sampling of the yeast proteome. Mol Cell Biol 19:7357–7368
Gygi SP, Rochon Y, Franza R, Aebersold R (1999) Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 19:1720–1730
Hartman D, Donald DR, Nikolaou S, Savin KW, Hasse D, Presidente PJA, Newton SE (2001) Analysis of developmentally regulated genes of the parasite Haemonchus contortus. Int J Parasitol 31:1236–1245
Henkle-Duhrsen K, Kampkotter A (2001) Antioxidant enzyme families in parasitic nematodes. Mol Biochem Parasitol 114:129–142
Kazura JW, Meshnick SR (1984) Scavenger enzymes and resistance to oxygen mediated damage in Trichinella spiralis. Mol Biochem Parasitol 10:1–10
Kotze AC (2003) Catalase induction protects Haemonchus contortus against hydrogen peroxide in vitro. Int J Parasitol 33:393–400
Kotze AC, McClure SJ (2001) Haemonchus contortus utilizes catalase in defence against exogenous hydrogen peroxide in vitro. Int J Parasitol 31:1563–1571
Liebau E, Eschbach M-L, Tawe W, Sommer A, Fischer P, Walter RD, Henkle-Duhrsen K (2000) Identification of a stress-responsive Onchocerca volvulus glutathione S-transferase (Ov-GST-3) by RT-PCR differential display. Mol Biochem Parasitol 109:101–110
McGonigle S, Dalton JP, James ER (1998) Peroxidoxins: a new antioxidant family. Parasitol Today 14:139–145
Mitsumoto A, Takanezawa Y, Okawa K, Iwamatsu A, Nakagawa Y (2001) Variants of peroxiredoxins expression in response to hydroperoxide stress. Free Radic Biol Med 30:625–635
Mkoji GM, Smith JM, Prichard RK (1988) Antioxidant systems in Schistosoma mansoni: correlation between susceptibility to oxidant killing and the levels of scavengers of hydrogen peroxide and oxygen free radicals. Int J Parasitol 18:661–666
Nare B, Smith JM, Prichard RK (1990) Schistosoma mansoni: levels of antioxidants and resistance to oxidants increases during development. Exp Parasitol 70:389–397
Ou X, Thomas GR, Chacon MR, Tang L, Selkirk ME (1995) Brugia malayi: differential susceptibility to and metabolism of hydrogen peroxide in adults and microfilariae. Exp Parasitol 80:530–540
Rachubinski RA, Subramani S (1995) How proteins penetrate peroxisomes. Cell 83:525–528
Reimer DL, Bailley J, Singh SM (1994) Complete cDNA and 5′ genomic sequences and multilevel regulation of the mouse catalase gene. Genomics 21:325–336
Selkirk ME, Smith VP, Thomas GR, Gounaris K (1998) Resistance of filarial nematode parasites to oxidative stress. Int J Parasitol 28:1315–1332
Singh AK, Shichi H (1998) A novel glutathione peroxidase in bovine eye. J Biol Chem 273:26171–26178
Smith NC (1989) The role of free oxygen radicals in the expulsion of primary infections of Nippostrongylus brasiliensis. Parasitol Res 75:423–438
Smith NC, Bryant C (1986) The role of host generated free radicals in helminth infections: Nippostrongylus brasiliensis and Nematospiroides dubius compared. Int J Parasitol 16:617–622
Tang L, Gounaris K, Griffiths C, Selkirk ME (1995) Heterologous expression and enzymatic properties of a selenium-independent glutathione peroxidase from the parasitic nematode Brugia pahangi. J Biol Chem 270:18313–18318
Tang L, Smith VP, Gounaris K, Selkirk ME (1996) Brugia pahangi: the cuticular glutathione peroxidase (gp29) protects heterologous membranes from lipid peroxidation. Exp Parasitol 82:329–332
Tripp C, Frank RS, Selkirk ME, Tang L, Grieve MM, Frank GR, Grieve RB (1998) Dirofilaria immitis: molecular cloning and expression of a cDNA encoding a selenium-independent secreted glutathione peroxidase. Exp Parasitol 88:43–50
Vernet P, Rigaudiere N, Ghyselinick N, Dufaure JP, Drevert JR (1996) In vitro expression of a mouse tissue specific glutathione peroxidase-like protein lacking the selenocysteine can protect stably transfected mammalian cells against oxidative stress. Biochem Cell Biol 74:125–131
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bagnall, N.H., Kotze, A.C. cDNA cloning and expression patterns of a peroxiredoxin, a catalase and a glutathione peroxidase from Haemonchus contortus. Parasitol Res 94, 283–289 (2004). https://doi.org/10.1007/s00436-004-1204-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-004-1204-7