
Abstract
To improve transfection efficiency in Trypanosoma cruzi, we developed a new electroporation protocol and expression vectors which use luciferase and green and red fluorescent proteins as reporter genes. In transient transfections, the electroporation conditions reported here resulted in luciferase expression 100 times higher than the levels obtained with previously described protocols. To verify whether sequences containing different trans-splicing signals influence reporter gene expression, we compared DNA fragments corresponding to 5′ untranslated plus intergenic (5′ UTR plus Ig) regions from GAPDH, TcP2β, α- and β-tubulin and amastin genes. Vectors containing sequences derived from the first four genes presented similar efficiencies and resulted in luciferase expression in transiently transfected epimastigotes that was up to 10 times higher than that for a control vector. In contrast, the amastin 5′ UTR plus Ig resulted in lower levels of reporter gene expression. We also constructed a vector containing an expression cassette designed to be targeted to the tubulin locus of the parasite.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The protozoan parasite Trypanosoma cruzi is the causative agent of Chagas’ disease, a debilitating disease endemic in many Latin American countries, where 16–18 million people are affected (World Health Organization 1999). Being part of a group that diverged early in eukaryotic evolution, T. cruzi, as well as other members of the kinetoplastid family, present many distinctive features regarding the mechanisms controlling gene expression. Some of these features include polycistronic transcription, trans-splicing processing of the pre-mRNA, mitochondrial RNA editing and transcription of a set of protein-coding genes carried on by RNA polymerase I (for a recent review see Clayton 2002). Since primary transcripts are polycistronic, cleavage of the pre-mRNA has to occur in the nucleus in order to produce monocistronic mRNAs. These cleavage reactions are linked to the addition of the 39-nucleotide miniexon [or spliced leader (SL)] at the 5′-end and the poly(A) tail at the 3′-end of each mRNA. SL addition results from a trans-esterification reaction called trans-splicing, which requires a conserved AG dinucleotide as SL addition site. Studies in various trypanosomatids have provided strong evidence demonstrating that SL addition and polyadenylation are not independent events. Instead, they are part of a “cut-and-paste” mechanism that occurs simultaneously or immediately after transcription; poly(A) selection is governed by the location of the SL-addition site of the downstream gene in the polycistronic primary transcript. In addition to the correct distance, the presence of a polypyrimidine-rich motif is also crucial, since only AG dinucleotides located downstream from a polypyrimidine tract are used as the SL acceptor site (Lebowitz et al. 1993; Matthews et al. 1994).
Most of the knowledge about the mechanisms of gene expression in these parasites resulted from the development of transfection protocols. In the first transfection experiments reported for T. cruzi, vectors containing a segment of the SL gene (Lu and Buck 1991) or the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene (Kelly et al. 1992) were placed upstream from the bacterial chloramphenicol acetyl transferase (CAT) gene. In the pTEX vector, described by Kelly et al. (1992), the Neo R selectable marker was also inserted. Although these earlier vectors contained all the sequences necessary for the expression of the exogenous mRNA, the lack of promoter elements resulted in relatively low levels of expression of the CAT gene. The introduction of sequences derived from rRNA promoter, as described by Teixeira et al. (1995), Tyler-Cross et al. (1995) and Martinez-Calvillo et al. (1997), resulted in a new generation of vectors, yielding expression of reporter genes at least two orders of magnitude greater in transfected epimastigotes. Besides the rRNA promoter, the only other promoter characterized in T. cruzi is the SL-RNA gene promoter, which yields only weak expression of reporter genes in epimastigotes (Nunes et al. 1997). Remarkably, transfection assays using promoter-trap plasmids and, more recently, genomic analyses have failed to reveal sequences with characteristics of the RNA polymerase II promoter in any trypanosomatid, even though transcription initiated by RNA polymerase II has been clearly described in these organisms.
To improve transfection efficiency in T. cruzi further, sequences placed downstream from the rRNA promoter should also be tested. These sequences may influence the correct processing of the pre-mRNA as well as the steady-state levels of the mature message. Various authors have described sequences derived from the 3′ untranslated region (UTR) plus intergenic (Ig) region of several genes, which contribute decisively to modulating gene expression in the parasite. (Nozaki and Cross 1995; Teixeira et al. 1995; Weston et al. 1999; Bartholomeu et al. 2003). In most cases, it has been demonstrated that these sequences are involved in mRNA stability control, but the presence of elements affecting the efficiency of polyadenylation must also be considered. On the other hand, the effect of sequences derived from the 5′ UTR and upstream Ig regions has been poorly investigated. These sequences may also contribute to mRNA stability and translation efficiency but, most importantly, they may contain signals determining trans-splicing efficiency. Here we evaluate the effect of various sequences containing the SL acceptor site and 5′ UTR derived from the surface protein amastin, α- and β-tubulin, the ribosomal protein TcP2β and GAPDH genes. These fragments were placed upstream from the luciferase reporter gene and tested in transiently transfected epimastigotes. In addition, we developed a vector that promotes the integration of an expression cassette into the tubulin locus of the parasite genome. This new vector allowed the generation of stable cell lines from transfected epimastigotes, which can be used in studies requiring differentiation into the other stages of the parasite’s life cycle.
Materials and methods
Parasite
Epimastigote forms of CL Brener and Colombiana (Col.1.7G2) clones and the Tulahuén strain of T. cruzi were grown at 28°C in liver infusion tryptose (LIT) medium supplemented with 10% fetal calf serum (FCS) as described by Camargo (1964).
Plasmids
All constructs used in transfection experiments were derived from the pGEM-luc vector (Promega). The pLR-Tub plasmid was generated by cloning the 600-bp XhoI-SacI 3′ UTR of α-tubulin gene fragment present in the pLATub vector (Bartholomeu et al. 2003) in the pGEM-luc vector downstream from the luciferase gene. A fragment amplified by the polymerase chain reaction (PCR) and containing the T. cruzi rRNA gene promoter was inserted upstream of luciferase. This fragment was generated by PCR amplification using total DNA purified from the CL Brener strain as template, with the forward primer (5′-CGGAGAAGCTTTTTGTAA-3′) and the reverse primer (5′-CTTGGGATCCCACGGTACCTTTGC-3′). The primer sequences were based on the analyses of the rRNA locus described by Dietrich et al. (1993). The forward primer has a HindIII site and the reverse primer has KpnI and BamHI sites. The 540-bp fragment corresponding to rRNA gene promoter was inserted into the HindIII and BamHI sites of pLR-Tub. The resulting plasmid contains two sites, KpnI and BamHI, between the rRNA promoter and the luciferase-coding region, which facilitate the insertion of the different 5′ UTR fragments.
The various 5′ UTR plus Ig regions were amplified using primers based on the sequences of α- and β-tubulin, GAPDH and TcP2β genes. To generate the plasmid pLR5′alphaTub, a 237-bp fragment containing the 5′ UTR plus Ig of T. cruzi α-tubulin gene was obtained by PCR amplification using total DNA from the Tulahuén strain as template with the forward primer (5′-CGCTGGGTACCGGGTGCACC-3′) and reverse primer (5′-AGAAGGATCCTATTTGAGGG-3′). The plasmid pLR5′betaTub was constructed by inserting into pLR-Tub a 304-bp fragment, which contained the 5′ UTR plus Ig of T. cruzi β-tubulin gene obtained by PCR amplification using total DNA from the Tulahuén strain as template with the forward primer (5′-GCCTGGTACCATCGTCCGCT-3′) and the reverse primer (5′-CTTGTCTGGATCCTGATGTT-3′). These primers contain KpnI and BamHI sites. The plasmid pLR5′gapdh was obtained by inserting a 450-bp fragment, derived from the 5′ upstream region of the T. cruzi gGAPDH I gene, into the KpnI and BamHI sites of the plasmid pLR-Tub. This fragment was isolated from the pTEX vector (Kelly et al. 1992) by digestion with SacI followed by treatment with T4 DNA polymerase. After this treatment, the same plasmid was digested with BamHI. The fragment obtained was inserted into the pLR-Tub vector previously digested with KpnI followed by treatment with T4 DNA polymerase and posterior BamHI digestion. To construct the plasmid pLR5′HX1, the fragment corresponding to the HX1 5′ upstream region of the TcP2β gene was obtained from pTREX (Vazquez and Levin 1999) after BamHI digestion. It was inserted into pLR-Tub, previously digested with BamHI. To generate the plasmid pLR5′Ama1, a modification in the vector pLRT previously described by Teixeira et al. (1995) was introduced by replacing the 3′ UTR plus Ig region from the TCR27 gene by a 600-bp XhoI-SacI fragment containing the 3′ UTR of the α-tubulin gene. The 3′ UTR plus Ig region of the α-tubulin gene fragment was generated by PCR amplification using total DNA from the Tulahuén strain as template and the forward primer (5′-AGTACTCGAGTGGCCGCTCCCGCGT-3′) and the reverse primer (5′-CGAGGAGCTCCACTAAATAGATCTT-3′). These primers have XhoI and SacI sites, respectively, to facilitate cloning (Bartholomeu et al. 2003). To ensure that all the plasmids used for the comparison of the influence of the 5′ sequences on reporter gene expression have the same vector background, we generated a second amastin plasmid, named pLR5′Ama2 by inserting a PCR fragment containing the amastin 5′ UTR from the pTCRGFPNeo vector (Teixeira et al. 1999a) into the BamHI site of the pLR-Tub vector. Sequences from all of these constructs were confirmed by automated sequencing using the MegaBace 1000 (Amersham-Pharmacia Biotech).
To create the pROCKGFPNeo vector, a β-tubulin cDNA cloned in pBluescript (Bartholomeu et al. 2003) was completely digested with EcoRI and partially with PstI. After filling in the EcoRI site with T4 DNA polymerase, the 1.0-kb tubulin fragment was cloned into the NotI/PstI digested pBluescript vector, generating pTUB plasmid. A cassette for foreign gene expression containing the neomycin resistance gene (Neo R) was obtained by KpnI digestion of the pTREX vector (Vazquez and Levin 1999), followed by filling the ends. This 3.1-kb fragment was cloned into pTUB, which was previously digested with HindIII/KpnI and treated with T4 DNA polymerase. A fragment containing the expression cassette plus the β-tubulin sequence upstream was obtained by SacI/NheI digestion of pTUB and was transferred to a modified pBluescript plasmid (pSk1) after digestion with NotI/KpnI and T4 DNA polymerase treatment. pSk1 does not carry the T7 and T3 promoters (LaCount et al. 2002). In this final construct we inserted into the XbaI/XhoI sites the GFP (green fluorescent protein) or the RFP (red fluorescence protein) genes, generating pROCKGFPNeo (Fig. 5A) or pROCKRFPNeo, respectively. The GFP coding sequence was derived from pTCRGFPNeo (Teixeira et al. 1999a) and the RFP coding sequence was derived from pDsRed vector (Clontech).
Electroporation
Epimastigotes growing at a density of 5×106–10×106 parasites/ml in LIT plus10% FCS were harvested, washed once with phosphate buffered saline (PBS) and resuspended at a density of 108 parasites/ml in the electroporation buffer (120 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4, 25 mM Hepes, 2 mM EDTA, 5 mM MgCl2, pH 7.6). Aliquots (0.4 ml) of cell suspension were mixed with 50 μl DNA (25, 50 or 100 μg) on ice cold 0.2-cm cuvettes and electroporated using a Bio-Rad gene pulser set at 0.3 kV and 500 μF, with two pulses (10 s between pulses). The time constants were always between 3.0 and 6.0 ms. For comparison, we also used the following protocol, which had been described by Ramirez et al. (2000): epimastigote cultures were harvested, washed once with PBS and resuspended to 2×108parasites/ml in electroporation buffer III (137 mM NaCl, 21 mM Hepes, 5 mM KCl, 5.5 mM Na2HPO4, 0.77 mM glucose, pH 7.0), and aliquots (0.7 ml) of cell suspension were mixed with 25 μg DNA in 0.4-cm cuvettes and electroporated using a Bio-Rad gene pulser set at 0.3 kV and 500 μF with two pulses. In both cases, the transfected cells were transferred to 5 ml LIT plus 10% FCS and incubated at 28°C for 48 h. All plasmid samples used in electroporation experiments were obtained by alkaline lysis using Qiagen columns (Qiagen).
Luciferase and β-galactosidase assays
Parasites were harvested 48 h after transfection, washed once with PBS, transferred to microtubes and centrifuged at 10,000 g for 10 s. Cell pellets were resuspended in 200 μl lysis buffer (100 mM potassium phosphate, pH 7.8, 0.4% Triton X-100 and 2 μg/ml leupeptin). After centrifugation for 1 min at 10,000 g, 4 μl of epimastigote lysates in the supernatants was diluted 1:5 and assayed as described previously (de Wet et al. 1987). In the transfection experiments using different 5′ UTR constructs, we used a plasmid containing the β-galactosidase reporter gene constructed by Coughlin et al. (2000) as a control plasmid, in order to avoid any possible artifact due to differences in transfection efficiency. In these cases, 10 μg control plasmid was co-transfected with each of the luciferase constructs and β-galactosidase assays were performed with 25 μl of the same cell lysate using O-nitrophenyl β-d-galactopyranoside, as described previously (Sambrook et al. 1989).
Flow cytometry analyses
Parasites (5×106) were washed with PBS plus 3% FCS and fixed for 30 min with MFF, freshly prepared by mixing equal volumes of solution I [1% (w/v) paraformaldehyde, 1% (w/v) sodium cacodylate, 0.67% (w/v) NaCl] with PBS. Fixed parasites were analyzed using a FACScan (Becton Dickinson), with 100,000 gated events acquired for analysis. Untransfected control cells, which show a significant amount of auto-fluorescence were used to standardize the parameters employed.
Pulse-field gel electrophoresis
Epimastigotes were included in agarose blocks as described by Engman et al. (1987). Pulse-field gel electrophoresis (PFGE) was carried out as reported by Cano et al. (1995) with the following modifications: the chromosomes were separated in 0.8% agarose gels using a program with 5 phases of homogeneous pulses (north/south, east/west) with interpolation for 135 h at 83 V. Phase 1 had pulse time of 90 s (run time 30 h); phase 2 200 s (30 h); phase 3 350 s (25 h); phase 4 500 s (25 h); phase 5 800 s (25 h). Chromosomes from Hansenula wingei (Bio-Rad) were used as molecular mass standards. Separated chromosomes were transferred to nylon filters and hybridized with 32-P labeled tubulin and GFP probes as described by Teixeira et al. (1994).
Results and discussion
Aiming to improve transient transfection efficiency of T. cruzi, we tested several published and a newly developed protocol based on the electroporation conditions used to transfect T. brucei and Leishmania. Here we compare our protocol with the electroporation conditions described by Ramirez et al. (2000), who used GFP as a reporter gene to test several parameters. Log phase cultures of epimastigotes were transfected with a construct carrying the luciferase gene driven by the rRNA promoter and containing 5′ and 3′ flanking sequences derived from amastin and α-tubulin genes, respectively. This vector, pLR5′Ama1, was derived from pLRT (Teixeira et al. 1995) from which the 3′ UTR plus Ig of the TCR27 gene were replaced by a fragment corresponding to the α-tubulin 3′ UTR and Ig. Previous experiments have shown that the presence of the α-tubulin 3′ UTR plus Ig resulted in luciferase expression in transfected epimastigotes that was 48% higher than in epimastigotes transfected with pLRT (Bartholomeu et al. 2003). As shown in Fig. 1, with our protocol we generated more than 18 times the luciferase activity achieved when the protocol described by Ramirez et al. (2000) was employed, using the same amount of DNA (25 μg). When we increased the DNA quantity up to 100 μg, the reporter activity was almost 100 times higher than before (Fig. 1). Thus, the presence of the α-tubulin 3′ UTR plus Ig, together with the electroporation conditions described here, resulted in a significant improvement in the efficiency of the transfection of epimastigotes.

Transfection efficiency in Trypanosoma cruzi cells. Epimastigote cultures of the CL Brener strain were transfected with different amounts of the pLR5′Ama1 plasmid. C25, C50 and C100 indicate luciferase activity in extracts of transfected parasites using 25, 50 and 100 μg of pLR5′Ama1, respectively, and the protocol described in “Material and methods”. R25 refers to the activity obtained with the protocol described by Ramirez et al. (2000) using 25 μg of the same plasmid. Data were derived from three independent experiments
Although the luciferase assays indicated that overall transfection efficiency had been improved, we prepared a plasmid vector containing the GFP reporter gene to determine the actual number of transfected parasites using our protocol. Using the pTREX vector (Vazquez and Levin 1999), we inserted the GFP coding region in the XbaI/XhoI sites generating the pTREXGFP plasmid. As shown in Fig. 2B, 24 h after transfection, 4% and 8% of cells transfected with 30 and 60 μg plasmid, respectively, were expressing high levels of GFP. These numbers are significantly higher than the transfection efficiency reported by Ramirez et al. (2000), who, after testing several strains, never observed a percentage of fluorescent parasites above 0.3% using 25 μg of a GFP-containing vector. Using pTREXGFP and the new transfection protocol, we were able to detect GFP-positive cells as soon as 12 h after transfection and even after 12 days in the absence of drug selection (Fig. 2B). Thus, it was possible to follow transiently transfected parasites for longer periods of time, in the absence of drug selection. To test whether the same efficiency of transient transfection can be achieved with a different reporter and also to verify whether a parasite can express two reporters at the same time using transient transfection assays, a similar plasmid was generated by replacing the GFP gene by the RFP coding region. Similarly to the expression of GFP, transfection of epimastigotes with 60 μg of the RFP plasmid (pTREXRFP) resulted in approximately 7% of positive parasites, detected 12 h post-transfection (not shown). As shown by the confocal microscopy analyses, parasites that were co-transfected with 30 μg of each GFP and RFP plasmids express high levels of green and red fluorescence distributed throughout the cell (Fig. 2A). The transfection efficiency with both markers is equivalent to the efficiency attained with each plasmid separately.

Transient expression of green fluorescent protein (GFP) and red fluorescent protein (RFP) in T. cruzi epimastigotes. Epimastigotes from the CL Brener strain were transfected with 30 or 60 μg of pTREXGFP or pTREXRFP, as described in “Material and methods”. A Differential interference contrast (DIC) and fluorescent images of epimastigotes expressing GFP and RFP, simultaneously, 24 h after co-transfection with 30 μg of each plasmid. These images were captured simultaneously on separate channels using a Zeiss LSM 510 laser scanning confocal microscope. B Kinetics of GFP expression after parasite transfection without drug selection, as determined using a fluorescent microscope. Each point corresponds to the percentage of green parasites in various fields with a total of 700 cells. Similar curves were obtained with parasite transfected with pTREXRFP and with both plasmids (not shown)
To test whether different 5′ UTR plus Ig regions may also influence the expression of a foreign gene in T. cruzi epimastigotes, we prepared the following constructs: the pLR5′alphaTub and pLR5′betaTub plasmids, which contain the 5′ UTR plus upstream sequences from T. cruzi α- and β-tubulin genes, respectively; the pLR5′gapdh plasmid with the sequence derived from the 5′ upstream region of the T. cruzi gGAPDH I gene; the pLR5′HX1 plasmid, which contains the fragment HX1 corresponding to the 5′ UTR and Ig region of the T. cruzi TcP2β gene (Vazquez and Levin 1999) and the pLR5′Ama2 plasmid in which the 5′ UTR plus Ig sequences from amastin gene were cloned upstream of luciferase in the same plasmid. In addition, we included in our experiments a plasmid pLucSL-control, which contains no T. cruzi sequences between the rRNA promoter and luciferase gene. Again, the 3′ UTR plus Ig region of α-tubulin was chosen to be placed downstream from luciferase in all constructs. The results of transient transfection experiments, shown in Fig. 3, demonstrated that the presence of sequences containing signals for addition of SL resulted in up to 10-fold increase in the levels of luciferase, as compared to the plasmid containing no sequences between the rRNA promoter and luciferase. Even though the control plasmid contains no upstream sequences directing trans-splicing of the luciferase mRNA, we detect significant levels of luciferase activity in parasites transfected with this plasmid. In three independent experiments, the mean value of luciferase activity in cells transfected with 50 μg of the control plasmid were close to 25,000 relative light units per106 cells. This result is in agreement with previous reports suggesting that the rRNA promoter sequence may contain elements that could potentially function as cryptic acceptor sites for trans-splicing reactions (Martinez-Calvillo et al. 1997).

The influence of 5′ untranslated region (UTR) sequences from different T. cruzi genes on transient gene expression. Different 5′ UTR sequences were cloned upstream from the luciferase reporter gene and 50 μg of each plasmid were used to transfect CL Brener epimastigotes using the protocol described in Fig. 1. The results are presented as the relative increases in luciferase activity compared to that of parasites transfected with a control plasmid, which has no T. cruzi sequences between the luciferase and the rRNA promoter. These results correspond to averages of three independent experiments, which were normalized using the values obtained from a co-transfected plasmid containing the β-galactosidase gene
Only small differences in relative luciferase activities were observed when cells were transiently transfected with four out of the five different plasmids (Fig. 3). The results of three independent experiments show that luciferase activities vary between 7- and 9-fold when sequences derived from α- and β- tubulin, TcP2β and GAPDH genes are compared. On the other hand, the construct containing the 5′ UTR plus Ig region of amastin genes is much less efficient, and resulted in luciferase levels that are only 3-fold higher than the control plasmid. In order to search for elements that could modulate the level of luciferase activity in the transfected parasites, we analyzed the different 5′ UTR and Ig sequences that were cloned upstream the luciferase gene. Polypyrimidine tracts present in the Ig regions act as bifunctional elements recognized by the processing machinery, which affect both polyadenylation and spliced-leader addition of the genes located upstream and downstream (Matthews et al. 1994; Schurch et al. 1994; revised by Vanhamme and Pays 1995). Deletion analyses and block substitution mutagenesis of the pyrimidine-rich sequence of α- and β- tubulin genes result in decreased expression of both CAT and luciferase reporter genes cloned upstream and downstream from this element (Matthews et al. 1994). In our experiments, a correlation between the polypyrimidine content present in the Ig regions and the values of luciferase activity can be observed if we compare the results obtained with the pLR5′Ama2 construct and the other four plasmids. As shown in Fig. 4, the 5′ flanking sequence of the amastin construct, which generated the lowest values of luciferase activity, presents the smallest number of consecutive pyrimidine residues.

The 5′ UTR sequences from different T. cruzi genes cloned upstream from the luciferase reporter gene. A α-Tubulin. B β-Tubulin. C gGAPDH I. D Ribosomal protein TcP2β. E Amastin. The different 5′ UTR sequences were cloned upstream from the luciferase coding region as described in “Material and Methods”. Highlighted sequences are polypyrimidine tracts; underlined sequences are potential AG splice-leader acceptor sites as determined by Teixeira et al. (1994) for the amastin gene, by Vazquez and Levin (1999) for TcP2β and by Bartholomeu et al. (2003) for α- and β-tubulin; and the doubly underlined codon is the luciferase start codon
In addition to the effect due to splicing signals, we must also consider the possibility that the sequences corresponding to the 5′ UTRs of these various genes may influence the fate of the luciferase mRNA by affecting its stability and/or the efficiency of translation. The transfection results are consistent with previous observations showing that among the genes from which the 5′ UTR sequences were tested, amastin is the only one whose expression is down regulated in epimastigotes as compared to amastigotes (Teixeira et al. 1995). However, for all genes analyzed here, the sizes of the 5′ UTR are extremely short, with an average of 46 nt between the translation start codon and the dinucleotide AG used as SL addition site (Fig. 4). There are only few reports in the literature describing regulatory elements in the 5′ UTR in trypanosomes, one being the study of the nuclear replication protein A and the kinetoplast topoisomerase II, from Crithidia fasciculata (Mahmood et al. 1999). Another study indicates that a fragment containing the 5′ UTR plus upstream Ig sequences derived from the low-abundance mRNA tuzin gene may contribute to suppress the expression of this multicopy gene by both inefficient RNA processing and poor translation initiation (Teixeira et al. 1999b). On the other hand, various groups have described the influence of 3′ UTR plus Ig sequences on T. cruzi gene expression. In their study, Nozaki and Cross (1995) suggested that the highest number of pyrimidine residues present in the Ig region of the GADPH gene or its structure could be responsible for the highest values of luciferase activity when compared to the corresponding region of the HSP60 and GP85 genes.
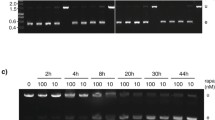
Finally, as an alternative approach to generating stable expression of transfected genes in T. cruzi, we constructed a new vector designed to allow integration by homologous recombination in the tubulin locus of the parasite’s genome. Only few reports have described the generation of stable transfected cell lines in T. cruzi using integrative vectors, which are transfected as linear plasmids containing sequences homologous to endogenous genes. In the case of pTCR27-2::Neo (Teixeira et al. 1999a), low efficiency results in generating stable transfectants were obtained, possibly because the target gene is a single copy gene. We decided to test whether targeting to a region containing a multicopy gene other than the rRNA could be used as a more efficient method to generate stable transfectants in T. cruzi. A vector containing an expression cassette flanked by β-tubulin sequences, named pROCKGFPNeo was constructed in such a way that any gene of interest can be inserted in place of GFP by double digestion with the enzymes XbaI/XhoI. To facilitate homologous recombination of tubulin sequences, the plasmid is linearized by NotI digestion, which has a unique site within the β-tubulin fragment (Fig. 5A). Using 50 μg of this vector, 0.4×108 epimastigotes of the Tulahuén strain were electroporated and selected in culture media containing 200 μg/ml G418. Drug selection for 4 weeks resulted in no parasites being detected in mock-transfected cultures and approximately 10% of G418-resistant parasites expressing GFP. To evaluate the stability of the GFP marker we performed analysis by fluorescence-activated cell sorter, which showed that 5 months after electroporation, the percentage of GFP-positive cells cultivated in the presence of 200 μg/ml G418 reached 40% (Fig. 5B, C). Moreover, after being cultivated for another 5 weeks in the absence of G418, 33% of the transfected population was still expressing high levels of GFP (Fig. 5D). To verify whether the GFP marker has been integrated in the tubulin locus, we analyzed a transformed population as well one isolated GFP-positive clone by pulse field gel electrophoresis. As shown on Fig. 6, Southern blot hybridizations indicate that GFP sequences were integrated in the tubulin cluster. Both GFP and β-tubulin probes hybridized with the same chromosomal band (approximately 3 Mb and 1.8 Mb in the Tulahuén and Col.1.7G2 strains, respectively), which were identified as the β-tubulin locus in the wild type parasites. However we cannot exclude the possibility that some of the plasmids were also maintained as episomal elements. We were able to transfect at least two different strains of T. cruzi with (Tulahuén and Col.1.7G2) and in both strains the construct has integrated in the predicted locus of the parasite genome. We have also determined that the cloned cell lines expressing GFP are infective in tissue culture cells (S. Pires, unpublished data). These and other pROCKGFPNeo transfected parasites are now available for biological studies in animal models of T.cruzi infections.

An integrative vector for stable expression of GFP in T. cruzi. A Schematic representation of the pROCKGFPNeo, a vector that was used for integration of an expression gene cassette at the β-tubulin locus. Cultures of epimastigote of the Tulahúen strain were transfected with 50 μg of the linearized vector and submitted to drug selection in the presence of 200 μg/ml of G418 for 6 months. After this period, one aliquot of the culture was kept in the absence of drug for 5 weeks. B Fluorescence-activated cell sorter (FACS) profile of non transfected cells growing in liver infusion tryptose (LIT) medium without drug. C Profile of G418-resistant populations expressing green fluorescent protein (GFP) 6 months after transfection. D Profile of the culture shown in C after cultivation for 5 weeks in the absence of drug selection

Integration of pROCKGFPNeo into the T. cruzi genome, showing chromosomal bands from wild-type epimastigotes (Wt) and from transfected cells (T) stained with ethidium bromide (EtBr) or hybridized with 32P-labeled probes for GFP and β−tubulin. In the left panel, the results with the Tulahuén strain, wild-type and a cloned, G418-resistant cell line generated after the transfection are shown. In the right panel we show the chromosomal separation and the hybridization results of wild type strain and transfected, G418-resistant population of the Col.1.7G2 T. cruzi cell line. The numbers on the right correspond to the sizes of a chromosomal molecular weight marker
References
Bartholomeu DC, Silva RA, Galvão LMC, El-Sayed N, Donelson JE, Teixeira SMR (2003) Trypanosoma cruzi: RNA structure and post-transcriptional control of tubulin gene expression. Exp Parasitol (in press)
Camargo EP (1964) Growth and differentiation in Trypanosoma cruzi: origin of trypanosomes in liquid medium. Rev Inst Med Trop São Paulo 6:93
Cano MI, Gruber A, Vazquez M, Cortés A, Levin MJ, González A, Degrave W, Rondinelli E, Zingales B, Ramirez JL, Alonso C, Requena JM, Silveira JF (1995) Molecular karyotype of clone CL Brener chosen for the Trypanosoma cruzi genome project. Mol Biochem Parasitol 71:273–278
Clayton CE (2002) Life without transcriptional control? From fly to man and back again. EMBO J 21:1881–1888
Coughlin BC, Teixeira SM, Kirchhoff LV, Donelson JE (2000) Amastin mRNA abundance in Trypanosoma cruzi is controlled by a 3′-untranslated region position-dependent cis-element and an untranslated region-binding protein. J Biol Chem 275:12051–12060
Dietrich P, Soares MB, Affonso MH, Floeter-Winter LM (1993) The Trypanosoma cruzi ribosomal RNA-encoding gene: analysis of promoter and upstream intergenic spacer sequences. Gene 125:103–107
Engman DM, Reddy LV, Donelson JE, Kirchhoff LV (1987) Trypanosoma cruzi exhibits inter- and intra-strain heterogeneity in karyotype and chromosomal gene location. Mol Biochem Parasitol 22:115–123
Kelly JM, Ward HM, Miles MA, Kendall G (1992) A shuttle vector which facilitates the expression of transfected genes in Trypanosoma cruzi and Leishmania. Nucleic Acids Res 20:3963–3969
LaCount DJ, Barrett B, Donelson JE (2002) Trypanosoma brucei FLA1 is required for flagellum attachment and cytokinesis. J Biol Chem 277:17580–17588
LeBowitz JH, Smith HQ, Rusche L, Beverley SM (1993) Coupling of poly(A) site selection and trans-splicing in Leishmania. Genes Dev 7:996
Lu HY, Buck GA (1991) Expression of an exogenous gene in Trypanosoma cruzi epimastigotes. Mol Biochem Parasitol 44:109–114
Mahmood R, Hines JC, Ray DS (1999) Identification of cis and trans elements involved in the cell cycle regulation of multiple genes in Crithidia fasciculata. Mol Cell Biol 19:6174–6182
Martinez-Calvillo S, Lopez I, Hernandez R (1997) pRIBOTEX expression vector: a pTEX derivative for a rapid selection of Trypanosoma cruzi transfectants. Gene 199:71–76
Matthews KR, Tschudi C, Ullu E (1994) A common pyrimidine-rich motif governs trans-splicing and polyadenylation of tubulin polycistronic pre-mRNA in trypanosomes. Genes Dev 8:491
Nozaki T, Cross GA (1995) Effects of 3′ untranslated and intergenic regions on gene expression in Trypanosoma cruzi. Mol Biochem Parasitol 75:55–67
Nunes LR, Carvalho MR, Shakarian AM, Buck GA (1997) The transcription promoter of the spliced leader gene from Trypanosoma cruzi. Gene 188:157–168
Ramirez MI, Yamauchi LM, de Freitas LH Jr, Uemura H, Schenkman S (2000) The use of the green fluorescent protein to monitor and improve transfection in Trypanosoma cruzi. Mol Biochem Parasitol 111:235–240
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
Schurch N, Hehl A, Vassella E, Braun R, Roditi I (1994) Accurate polyadenylation of procyclin mRNAs in Trypanosoma brucei is determined by pyrimidine-rich elements in the intergenic regions. Mol Cell Biol 14:3668–3675
Teixeira SMR, Russell DG, Kirchhoff LV, Donelson JE (1994) A differentially expressed gene family encoding “amastin”, a surface glycoprotein of Trypanosoma cruzi amastigotes. J Biol Chem 269:20509
Teixeira SM, Kirchhoff LV, Donelson JE (1995) Post-transcriptional elements regulating expression of mRNAs from the amastin/tuzin gene cluster of Trypanosoma cruzi. J Biol Chem 270:22586–22594
Teixeira SMR, Otsu K, Hill KL, Kirchhoff LV, Donelson JE (1999a) Expression of a marker for intracellular Trypanosoma cruzi amastigotes in extracellular spheromastigotes. Mol Biochem Parasitol 98:265–270
Teixeira SM, Kirchhoff LV, Donelson JE (1999b) Trypanosoma cruzi: suppression of tuzin gene expression by its 5′-UTR and spliced leader addition site. Exp Parasitol 93:143–151
Tyler-Cross RE, Short SL, Floeter-Winter LM, Buck GA (1995) Transient expression mediated by the Trypanosoma cruzi rRNA promoter. Mol Biochem Parasitol 72:23–31
Vanhamme L, Pays E (1995) Control of gene expression in trypanosomes. Microbiol Rev 59:223–240
Vazquez MP, Levin MJ (1999) Functional analysis of the intergenic regions of TcP2β gene loci allowed the construction of an improved Trypanosoma cruzi expression vector. Gene 239:217–225
Weston D, La Flamme AC, Van Voorhis WC (1999) Expression of Trypanosoma cruzi surface antigen FL-160 is controlled by elements in the 3′ untranslated, the 3′ intergenic, and the coding regions. Mol Biochem Parasitol 102:53–66
Wet JR de, Wood KV, DeLuca M, Helinski DR, Subramani S (1987) Firefly luciferase gene: structure and expression in mammalian cells. Mol Cell Biol 7:725–737
World Health Organization (1999) The World Health Report. http://www.who.ch. Cited 9 May 2003
Acknowledgements
The authors are grateful to Dr. John E. Donelson from the University of Iowa, Iowa, USA for providing a training opportunity for WDR and valuable suggestions, Etel R. Vieira for FACS analyses, and to Alice Machado-Silva for critical reading of the manuscript. We are also thankful to Dr. Egler Chiari for providing some of the T. cruzi strains. This work was supported by funds from the World Health Organization/Special Program for Research and Training in Tropical Diseases (WHO/TDR) and the NIH Fogarty International Research Collaborative Award (FIRCA). The work of WDR, RAS, DCB, SFP and SMRT received further support from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brazil. The work of MV and MJL was supported by grants from FONCYT-PICTs 01-05225 and 01-06803 TWAS research grants 00–311 RG/BIO/LA. In addition, MJL was supported by an International Research Scholar grant from the Howard Hughes Medical Institute, Chevy Chase, Md., USA. The experiments described here were performed in Brazil and comply with the current laws of the country.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
DaRocha, W.D., Silva, R.A., Bartholomeu, D.C. et al. Expression of exogenous genes in Trypanosoma cruzi: improving vectors and electroporation protocols. Parasitol Res 92, 113–120 (2004). https://doi.org/10.1007/s00436-003-1004-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-003-1004-5