
Abstract
CD47, a transmembrane protein, acts as a “do not eat me” signal that is overexpressed in many tumor cell types, thereby forming a signaling axis with its ligand signal regulatory protein alpha (SIRPα) and enabling the tumor cells to escape from macrophage-mediated phagocytosis. Several clinical trials with CD47 targeting agents are underway and have achieved impressive results preliminarily. However, hematotoxicity (particularly anemia) has emerged as the most common side effect that cannot be neglected. In the development of CD47 targeting agents, various methods have been used to mitigate this toxicity. In this review, we summarized five strategies used to alleviate CD47 blockade-induced hematotoxicity, as follows: change in the mode of administration; dual targeting bispecific antibodies of CD47; CD47 antibodies/SIRPα fusion proteins with negligible red blood cell binding; anti-SIRPα antibodies; and glutaminyl-peptide cyclotransferase like inhibitors. With these strategies, the development of CD47 targeting agents can be improved.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immunotherapy, particularly targeting with immune checkpoint inhibitors, is the most significant advancement in antitumor therapy (Sharma and Allison 2015). Blocking T cell inhibitory pathways (such as CTLA-4, PD-1, PD-L1, and others) to activate effector T cell and enhance antitumor immune responses has been successfully used in clinic. However, due to the low response rate, the majority of cancer patients still cannot benefit from them. Besides, many patients who initially responded to these treatments ultimately developed drug resistance and progressive disease (Ribas and Wolchok 2018). Although research works focused on promoting adaptive immune antitumor responses, an increasing number of studies are exploring approaches that activate innate immune cells, such as macrophages. Macrophages engulf and eliminate tumor cells, and this process is known as phagocytosis (Freeman and Grinstein 2014). After phagocytosis, macrophages can also activate T cells by presenting antigen and bridging innate and adaptive immunity. CD47, a glycosylated five-transmembrane protein, also known as integrin associated protein, was recently found to be involved in a range of cellular process, including apoptosis, proliferation, adhesion and migration (Liu et al. 2020). Furthermore, CD47 functions as a “don’t eat me” signal and is highly expressed in many tumors. After the extracellular segment of CD47 combines with the ligand signal regulatory protein alpha (SIRPα) expressed on macrophages, the immunoreceptor tyrosine-based inhibitory motifs in cytoplasmic domain of SIRPα are phosphorylated. Then the Src homology region 2-domain-containing phosphatase-1 and -2 are recruited and activated. Finally, tumor cells send inhibitory signals to macrophages and prevent phagocytosis and tumor antigen presentation (McCracken et al. 2015; Feng et al. 2019). Therefore, targeting CD47 is a new and important strategy in antitumor immunotherapy.
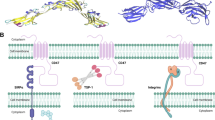
Hu5F9-G4 (magrolimab) is the first CD47 antibody which has entered clinical development and produced encouraging results preliminarily. Of the patients with relapsed or refractory non-Hodgkin’s lymphoma, 50% had objective response, and 36% had complete response (Advani et al. 2018). In myelodysplastic syndrome, magrolimab in combination with azacitidine had 91% objective response, with 42% achieving complete remission (Sallman et al. 2019). Based on this, the FDA granted breakthrough therapy designation for magrolimab. Recently, the SIRPα-IgG1 Fc, TTI-621 also exhibited promising prospects in patients with relapsed or refractory hematologic malignancies; for DLBLC patients, the objective response rate was determined to be 29% (Ansell et al. 2021). Moreover, several other approaches targeting the CD47/SIRPα pathway were implemented. At present, more than 20 CD47/SIRPα targeting agents (Table 1) have proceeded to active phase I/II/III clinical trials, which involved hematological malignancies and solid tumors. However, with the release of safety profiles, anemia has emerged as the most common dose-related toxicity when these agents were used as monotherapy or in combination, exactly as preclinical studies indicated (Buatois et al. 2018; Liu et al. 2015; Olsson and Oldenborg 2008). Mechanistically, CD47 is overexpressed in tumor cells (Willingham et al. 2012; Jaiswal et al. 2009), and expressed in normal cells, such as red blood cells (RBCs) and blood platelets; it represents a self-signal to protect normal cells from phagocytosis by macrophages (Oldenborg et al. 2000). Blocking CD47 would result in macrophage engulfing the RBCs. Moreover, administration of CD47 antibodies targeting RBCs induced antibody-dependent cellular phagocytosis (ADCP). A reaction known as hemagglutination also plays an important role in CD47 antibody-induced anemia. Hemagglutination is a form of RBCs agglutination that is caused by antibodies, lectins, and viral proteins (Muramatsu et al. 2014; Rizzo et al. 2014). In the presence of CD47 antibody, random binding to CD47 could occur on the membrane of different RBCs, thereby further accelerating their clearance (Fernandes et al. 2011) (Fig. 1). Therefore, balancing the improvement of antitumor efficacy and mitigating the potential toxicity of hematology, especially anemia, is among the critical concerns in the development and clinical application of CD47 targeting agents. In this review, we summarized and discussed the five most promising strategies to diminish CD47 blockade-induced hematotoxicity.

Anti-CD47 antibodies induce hematotoxicity. A Anti-CD47 antibodies activate phagocytosis of RBCs by macrophages. B Anti-CD47 antibodies bind to different RBCs to cause RBC agglutination. Ab antibody, MΦ macrophage
The change in the mode of administration
A low priming dose of CD47 antibody, which induces compensatory reticulocytosis (an increase in reticulocytes due to the increase in marrow activity) to replace RBCs loss, could effectively alleviate anemia (Fig. 2A). Hu5F9-G4, a CD47-blocking monoclonal antibody with humanized IgG4, has been successfully applied at a low priming dose of 1 mg/kg and a higher maintenance dose of 30–45 mg/kg to mitigate anemia. In preclinical toxicology studies, the major dose-limiting toxicity was anemia; this could be alleviated using a low priming dose and a higher maintenance dose. The results indicated that non-human primates could tolerate Hu5F9-G4 doses up to 300 mg/kg, but no maximum tolerated dose (MTD) was reached (Liu et al. 2015). In a phase I clinical trial that evaluated safety, pharmacokinetics, and pharmacodynamics, Hu5F9-G4 was used alone in patients with advanced cancers, including solid tumors and lymphoma. No MTD was reached in 62 patients. Hematologic toxicities occurred as follows without apparent clinical consequences: transient anemia (fall in hemoglobin 1–2 g/dL) in 57% of patients; and hemagglutination on peripheral blood smear in 36% of patients (Sikic et al. 2019). In another phase Ib clinical trial, the use of Hu5F9-G4 synergizing with rituximab for the treatment of relapsed or refractory non-Hodgkin’s lymphoma resulted in an objective response rate of 50% with 36% complete response, this administration caused predictable and transient mild anemia of grade 1 or 2 primarily in the first week of the priming dose (Advani et al. 2018). The results of a phase Ib clinical trial to assess tolerability and efficacy of magrolimab plus azacytidine in myelodysplastic syndrome and acute myeloid leukemia patients also indicated that the safety of the combo was similar to that of azacytidine alone (Sallman et al. 2020). Moreover, a randomized, double-blind, placebo-controlled multicenter phase III study is being conducted to compare the effects of treatment with magrolimab plus azacitidine and placebo plus azacitidine in untreated patients with myelodysplastic syndrome; results would further confirm efficacy and safety of the treatment (NCT04313881).

Strategies to diminish CD47 blockade induced hematotoxicity. A The priming low dose of CD47 antibody is administered to eliminate aged RBCs and lead to compensatory of reticulocytes. A higher maintenance dose is administered for better treatment. B Dual-targeting bispecific antibodies that target both tumor-associated antigen and CD47 can reduce RBCs binding while ensuring the binding to tumor cells. C CD47 antibodies/SIRPα fusion proteins with negligible RBC binding can reduce hematotoxicity. D Anti-SIRPα antibody does not eliminate RBCs while blocking the CD47/SIRPα pathway. E QPCTL inhibitors block the CD47/SIRPα pathway by altering the post-translational modification of CD47 and do not cause hematotoxicity. Ab antibody, MΦ macrophage, TAA tumor-associated antigen, Fp fusion protein, w/o pGlu without pyroglutamate modification
Mechanistically, a lower priming dose leads to prior clearance of aged RBCs by macrophages. However, rapid compensatory reticulocytosis immediately follows, and younger RBCs are released into the blood circulation. These young RBCs do not express significant pro-phagocytic signals that have accumulated on older RBCs and are thus less affected by Hu5F9-G4 (Arias and Arias 2017). Therefore, despite higher continued Hu5F9-G4 dosing, hemoglobin levels remained stable or declined slightly after the first week. Moreover, broadly expressed CD47 in RBCs indicates that large doses or frequent administration may be required, i.e., the so-called “antigen sink” effect and may promote the metabolization of CD47 antibody. The phase I clinical trial data of Hu5F9-G4 showed that even the dose of 10 mg/kg also exhibited nonlinear PK, indicating that the administration of a high dose was necessary to achieve 100% receptor occupancy (Agoram et al. 2018). In most patients, anemia gradually alleviated and the level of hemoglobin returned to baseline, as the circulating RBCs shifted to a younger age. Moreover, a novel process called RBC pruning may also illustrate the schedule of administration. The initial lower priming dose of Hu5F9-G4 rapidly reduced CD47 expression on the surface of RBCs, and then these pruned RBCs were unaffected by subsequent Hu5F9-G4 dosing. However, the specific mechanism is unclear and needs to be further studied (Chen et al. 2018).
Dual targeting bispecific antibodies of CD47
Dual targeting bispecific antibodies can be designed to target both tumor-associated antigens (TAA) and CD47, thereby reducing the binding of CD47 expressed in normal cells (Fig. 2B). TAA and CD47 bispecific antibodies with Fc-dependent killing function, can effectively kill tumor cells and prevent damage on erythrocytes that express CD47, thereby improving the safety of CD47 blocking therapy.
CD19 is a B cell-specific marker and is widely expressed in B cell malignancies (Katz and Herishanu 2014). TG-1801, a CD47/CD19 bispecific antibody with humanized IgG1 developed by TG Therapeutics, is now undergoing phase I clinical trial in B cell lymphoma (NCT03804996). Dheilly et al. (2017) developed a CD47/CD19 bispecific antibody that showed higher binding affinity to B cell lymphoma than CD47 monoclonal antibody. The better efficacy of CD47/CD19 bispecific antibody in phagocytosis experiments was demonstrated using human macrophages and in vivo experiments in mice. Even at the highest concentration of 150 μg/mL, the CD47/CD19 bispecific antibody rarely bound to erythrocytes. Incubation of the bispecific antibody with whole blood showed that the plasma concentration of the antibody was not significantly reduced by cell adsorption, confirming the low possibility of interacting with human erythrocytes. The antibody also maintained its blood concentration instead of being cleared quickly in cynomolgus monkeys. The latter studies further indicated that the appropriate weakening of the binding capacity of the CD47/CD19 bispecific antibody to CD47 could reduce the binding to CD19 negative cells and improve the safety of the bispecific antibody (Buatois et al. 2018; Dheilly et al. 2018). To be specific, the CD47/CD19 bispecific antibody that weakly binds to CD47 showed less binding to mouse erythrocytes in vitro and exhibited less effect on mouse RBCs value in vivo. In cynomolgus monkeys, the CD47/CD19 bispecific antibody with strong and weak CD47 binding ability had clearance values of 18.24 and 12.24 mL/day/kg, respectively. The bispecific antibody with weak CD47 binging ability also showed a higher initial serum concentration.
Anti-CD47/CD20 is another type of bispecific antibody that is used in the treatment of B cell lymphoma, it corresponds to the clinical application of anti-CD20 monoclonal antibody rituximab (Mohammed et al. 2019). IMM0306, a CD47/CD20 bispecific antibody with IgG1 developed by ImmuneOnco, is now undergoing phase I clinical trial in lymphoma (CXSL1900097). Piccione et al. (2015) reported that the CD47/CD20 bispecific antibody selectively bound to dual positive B lymphoma cells, promoted phagocytosis of various B lymphoma cells by macrophages and inhibited tumor growth in mouse. When CD20 positive tumor cells are mixed with excess erythrocytes and incubated with the bispecific antibodies, the antibodies selectively bind to the tumor cells, but not to the erythrocytes. A similar design is shown in the fusion of the SIRPα protein targeting CD47 with the rituximab targeting CD20 to generate a bispecific antibody, that is selectively bound to tumor cells and to reduce the tumor burden in mouse xenograft lymphoma models (Piccione et al. 2016). Binding to erythrocytes of human or cynomolgus monkey was not detected at a bispecific antibody concentration of up to 500 μg/mL in vitro. The single dose of 3, 10, or 30 mg/kg bispecific antibody in cynomolgus monkey did not cause the obvious reduction of erythrocytes and hemoglobin within 2 weeks. Ma et al. (2020) developed a CD47/CD20 bispecific antibody consisting of anti-CD47 nanobody and rituximab. This bispecific antibody showed more preferential binding to lymphoma cells in the competition binding assay that involved Raji cells and erythrocytes.
In addition, CD47 bispecific antibodies have been studied in solid tumors. The CD47/EGFR bispecific antibody could target human epidermoid carcinoma A431 cells, thereby promoting the phagocytosis of macrophages and improving the treatment in xenograft tumor models (Yang et al. 2018). Meanwhile, the significant reduction of RBCs was not obvious in mice after CD47/EGFR bispecific antibody treatment at a dose of 10 or 40 mg/kg. The dual-targeting fusion proteins formed with PD-L1 antibody and SIRPα recombinant protein can target CD47 and PD-L1, simultaneously blocking the two inhibitory signaling pathways of CD47/SIRPα in innate immune and PD-1/PD-L1 in adaptive immunity. The dual-targeting fusion proteins could bind to several solid tumor cells and inhibit the growth of colon cancer MC38 cells in the mouse tumor model (Liu et al. 2018a,b). In terms of safety, the dual-targeting fusion proteins showed weak binding to erythrocytes in vitro. Thus, it did not induce a significant decrease in erythrocytes and hemoglobin, thereby maintaining blood concentrations in mice. Shi et al. (2020) designed the humanized CD47 antibody h4C1. Based on this, the CD47/PD-L1 bispecific antibody was further constructed; it displayed safety in hemagglutination assay, which is an in vitro assay that reflects in vivo hematotoxicity. More specifically, the RBCs that link together form a diffuse lattice instead of precipitating at the bottom of the container to form a red dot in the assay (Killian 2014). Compared with the high concentration of h4C1 which induced agglutination of human erythrocytes, the bispecific antibody did not cause hemagglutination at the concentration range of 5.65 ng/mL to 1 mg/mL (Shi et al. 2020). Innovent Biologics developed the CD47/PD-L1 bispecific antibody—IBI322, and this antibody is now undergoing phase I clinical trials (NCT04338659 and NCT04328831). In non-human primate studies, RBCs reduction rate was less than 20% when IBI322 was used at 10 mg/kg. However, the same dose of Hu5F9 led to a 35% reduction, which indicated that IBI322 had lower hematotoxicity than Hu5F9 (Wang et al. 2020a).
For dual targeting bispecific antibodies of CD47, some points are worth noting. The weak binding of the bispecific antibody to CD47 seems to be important for reducing hematotoxicity, but whether it reduces the efficacy of the drug or not should be considered. In addition, the differential affinity between the CD47-binding arm and the TAA-binding arm that enables the bispecific antibody to preferentially bind to TAA to achieve specific phagocytic function still needs further study. Therefore, maintaining the balance of the binding ability of bispecific antibodies to CD47 during drug development is necessary. Whether bispecific antibodies cause more side effects due to dual targeting needs further elucidation.
CD47 antibodies/SIRPα fusion proteins with negligible RBC binding
Some CD47 antibodies can avoid erythrocyte binding and selectively bind to tumor cells, thereby reducing hematotoxicity (Fig. 2C). TJC4 (TJ011133) is a CD47 antibody developed by I-Mab that has differential erythrocytes binding property. It is a fully human monoclonal antibody with IgG4; it is undergoing phase I/II clinical trials in acute myeloid leukemia (NCT04202003) and phase I clinical trials in lymphoma and solid tumors (NCT03934814). Scientists of I-Mab screened CD47 binders from phage libraries and transformed them into antibodies. Through a series of functional experiments, TJC4, which has minimal erythrocytes binding ability and does not cause hemagglutination, was selected (Meng et al. 2019). TJC4 enhances the macrophage phagocytosis of Raji cells and human primary acute myeloid leukemia cells. The studies in mouse xenograft models of Raji B cell lymphoma, HL60 leukemia, WSU-DLCL2 DLBCL, and patient-derived liver cancer showed that TJC4 had antitumor ability in vivo. TJC4 showed weak binding of human erythrocytes, and the concentrations in the range of 0.003–100 μg/mL did not cause hemagglutination. In cynomolgus monkeys, a single intravenous injection of TJC4 at 15 mg/kg did not induce an obvious reduction of erythrocytes, hemoglobin and platelets. Even 10–30 mg/kg TJC4 intravenous injection (once per week for five times) also showed minimal decreases in erythrocytes and hemoglobin (Meng et al. 2019).
AO-176 is another CD47 antibody that can preferentially bind to tumor cells. It is a humanized monoclonal antibody with IgG2 and is developed by Arch Oncology. AO-176 is undergoing phase I clinical trial in solid tumors (NCT03834948). Puro et al. (2020) reported that AO-176 promoted the phagocytosis of human macrophages to several solid and hematologic tumor cells, thereby inducing the apoptosis of tumor cells. Findings showed the in vivo antitumor capability of AO-176 in mouse tumor models. In terms of safety, the binding ability of AO-176 to human normal cells expressing CD47 was weaker than the binding ability to the tumor cells. Binding to RBCs was particularly negligible. AO-176 at 0.017–1000 μg/mL did not cause hemagglutination in vitro. In cynomolgus monkeys, the RBCs and hemoglobin values were stable after the intravenous injection of AO-176 (Puro et al. 2020). Moreover, the affinity of AO-176 to CD47 under acidic conditions at pH 6.5 was much stronger than that at physiological pH (Bouchlaka et al. 2018). The pH-dependent binding of AO-176 may enhance its targeting to tumor cells rather than erythrocytes in vivo due to the acidic conditions in the solid tumors microenvironment.
Similar to the CD47 antibody, some SIRPα fusion proteins that target CD47 can also distinguish tumor cells from erythrocytes. TTI-621, developed by Trillium, is a fusion protein of the CD47-binding domain of wild type SIRPα and human IgG1 which is undergoing phase I clinical trials in hematologic malignancies and solid tumors (NCT02663518 and NCT02890368). According to the early clinical trial results, TTI-621 showed 36% objective response rate in advanced DLBCL and reduced tumor burden in Sézary syndrome (a leukemic variant of cutaneous T-cell lymphoma) (Ansell et al. 2017; Johnson et al. 2019). Preclinical experiments results indicated that TTI-621 promoted the phagocytosis of various human hematological or solid tumor cell lines by macrophages, and the phagocytosis of primary blood tumor cells (Petrova et al. 2017). The in vivo experiments demonstrated that TTI-621 inhibited acute myeloid leukemia and B-cell lymphoma in mouse tumor models. Although TTI-621 bound to cynomolgus monkey's erythrocytes and caused anemia in monkey, it was safe to human erythrocytes. TTI-621’s binding ability to human erythrocytes was weak, and 0.04–2000 nmol/L of TTI-621 did not cause human hemagglutination (Petrova et al. 2017). Stable hemoglobin levels and transient thrombocytopenia without clinical sequelae were observed in patients infused with TTI-621 (Ansell et al. 2016, 2017). IMM01, a SIRPα and IgG1 fusion protein developed by ImmuneOnco, also has minimal binding to the CD47 of erythrocytes and is undergoing phase I clinical trials in lymphoma (CTR20191531).
Regarding the mechanism of selective binding of the CD47 antibodies or SIRPα fusion protein to tumor cells and normal cells, TJC4 was able to recognize a unique CD47 epitope through the crystal structure analysis. This binding epitope was masked on erythrocytes due to glycosylation modification and could not be fully exposed; thus, TJC4 did not bind to erythrocytes (Meng et al. 2019). Multiple sites in CD47 extracellular IgV domain can be modified by glycosylation, which affects its function (Lindberg et al. 1993; Kaur et al. 2011). Some differences in CD47 glycosylation may be present in different cells, which results in the selective binding of CD47 antibodies. Another hypothesis of the differential binding property of CD47 antibodies is as follows. The membrane mobility of erythrocytes CD47 is reduced because of its association with the spectrin cytoskeleton; thus, CD47 cannot be effectively clustered, thereby resulting in the ineffective binding of the antibodies to erythrocytes CD47 (Mouro-Chanteloup et al. 2003; Subramanian et al. 2006). In addition, CD47 possibly has different binding partners or interacting proteins in different cells, which may affect the antibody binding epitopes and results in binding differences (Bruce et al. 2002; Barazi et al. 2002; Velliquette et al. 2019). However, current studies on the mechanisms underlying the differential binding property of CD47 antibodies are still insufficient. At present, the development of CD47 antibodies/SIRPα fusion proteins with negligible RBC binding is only based on the large-scale screening. Further study of the specific difference of CD47 between tumor cells and normal cells is still needed to guide the development of CD47 targeting reagents.
Anti-SIRPα antibodies
Antibodies that target the CD47 receptor-SIRPα can be used as a strategy to eliminate hematotoxicity (Fig. 2D). Anti-SIRPα antibodies do not target erythrocytes and therefore do not cause hemagglutination (Voets et al. 2019). OSE-172 (BI 765063) is an anti-SIRPα antibody developed by OSE Immunotherapeutics that is now undergoing phase I clinical trials in solid tumors in cooperation with Boehringer Ingelheim (NCT03990233). Moreover, the anti-SIRPα antibody CC-95251 developed by Celgene is undergoing phase I clinical trial in the solid and hematologic tumors (NCT03783403), and the anti-SIRPα antibody FSI-189 developed by Gilead Sciences is undergoing phase I clinical trials in non-Hodgkin’s lymphoma (NCT04502706). For the effectiveness of targeting SIRPα, the anti-SIRPα antibody ADU-1805 enhanced the macrophages phagocytosis and neutrophil trogocytosis [a process neutrophils kill tumor cells (Matlung et al. 2018)] on Raji cells when combined with rituximab (Voets et al. 2019). ADU-1805 did not bind human erythrocytes and platelets at concentration up to 100 nM and did not cause hemagglutination at concentrations in the range of 0.1–400 nM. Pharmacokinetic studies in cynomolgus monkeys indicated that intravenous administration of 0.3–30 mg of ADU-1805 had the estimated half-life of 1.86–6.41 days, and did not obviously affect the hemoglobin levels of male and female cynomolgus monkeys. However, considering that ADU-1805 is an anti-human SIRPα antibody, and SIRPα is expressed on immune cells, ADU-1805 still lacks the validity of antitumor animal experiments due to the species matching problem of the antibody and target. The results of experiments on mouse anti-SIRPα antibody MY-1 provided supporting evidence for the effectiveness and safety of anti-SIRPα antibody. MY-1 combined with rituximab significantly inhibited tumor growth in Raji tumor model mice (Yanagita et al. 2017). In addition, intraperitoneal injection of MY-1 in C57BL/6 mice had no obvious effect on RBCs, hemoglobin and platelets, suggesting its safety.
The administration of anti-SIRPα antibodies to syngeneic mouse tumor models could significantly inhibit the growth of renal cell carcinoma RENCA cells and the metastasis of melanoma B16BL6 cells (Yanagita et al. 2017), and also reduce the tumor growth of breast cancer 4T1, mesothelioma AK-7 and colon cancer MC38 cells (Durand et al. 2018). However, anti-SIRPα antibody treatment alone could not significantly increase the phagocytosis of Raji, breast cancer SK-BR-3 and colon cancer DLD-1 cells by human macrophages and could not inhibit the growth of Raji and CT26 cells in mice (Yanagita et al. 2017; Ring et al. 2017; Sim et al. 2019). This finding is due to the fact that the CD47 antibody targets tumor cells whereas the anti-SIRPα antibody targets immune cells, which usually does not induce the antibody-dependent cell-mediated cytotoxicity (ADCC) and ADCP against tumor cells. The effectiveness of anti-SIRPα antibody alone in RENCA and B16BL6 cells is due to the high expression of SIRPα in renal cell carcinoma and melanoma (Yanagita et al. 2017). Based on this result, the application of anti-SIRPα antibody alone in other tumors is limited. One solution to this problem is to combine anti-SIRPα antibodies with antibodies that target TAA. The combination of anti-SIRPα and anti-CD20 rituximab could significantly promote the phagocytosis of Raji cells by macrophages and inhibit the growth of Raji cells in mice. The combinations of anti-SIRPα and anti-HER2 trastuzumab or anti-EGFR cetuximab were also effective in breast cancer SK-BR-3 or colon cancer DLD-1 cells (Voets et al. 2019; Yanagita et al. 2017; Ring et al. 2017; Sim et al. 2019). Another solution is to activate adaptive immunity along with the activation of innate immunity using anti-SIRPα antibodies. The combination of anti-SIRPα and anti-PD-1 or anti-4-1BB showed antitumor ability in several syngeneic mouse tumor models (Yanagita et al. 2017; Durand et al. 2018).
In addition, adaptability and specificity are critical for anti-SIRPα antibodies. The extracellular IgV domain of SIRPα that binds to CD47 is highly polymorphic. Human SIRPα has two allelic variants, namely, v1 and v2 (Sim et al. 2019). The ability of various anti-SIRPα antibodies to bind to both v1 and v2 variants indicates that the current development of anti-SIRPα antibodies has considered adaptability (Voets et al. 2019; Ring et al. 2017; Sim et al. 2019). SIRPα has a high degree of homology with SIRPβ and SIRPγ. Both SIRPα and SIRPβ are expressed in myeloid cells, whereas SIRPγ is expressed in T, NK, and NKT cells (Voets et al. 2019; Sim et al. 2019). Non-selective targeting of SIRPα and SIRPγ reportedly impairs the normal function of human T cells (Gauttier et al. 2020). Therefore, anti-SIRPα has higher requirements in the specificity of antibodies that refrain from binding to SIRPβ and SIRPγ. Researchers who developed anti-SIRPα antibodies considered their specificity. For example, ADU-1805 showed stronger binding ability with SIRPα than SIRPβ and SIRPγ (Voets et al. 2019), and OSE-172 had little effect on the function of T cells because it did not bind to SIRPγ (Gauttier et al. 2018).
Glutaminyl-peptide cyclotransferase like (QPCTL) inhibitors
QPCTL inhibitors (SEN177 and PQ912) represent another promising approach for targeting CD47-SIRPα axis with good efficacy and tolerance (Fig. 2E). Several studies showed that QPCTL is an enzyme used to catalyze the cyclization of N-terminal glutamine and glutamic acid residues on CD47 into an N-terminal pyroglutamate residue (Logtenberg et al. 2019; Mair et al. 2019; Wu et al. 2019). High expression of QPCTL is associated with poor prognosis of patients with renal cancer and leukemia (Wu et al. 2019).
Logtenberg et al. (2019) reported that deletion or inhibition of QPCTL limited the signaling transduction of CD47-SIRPα axis and enhanced the phagocytosis capacity of macrophages to eliminate tumor cells. In vitro phagocytosis assays indicated that SEN177 treatment could increase the clearance of Raji cells. Similarly, SEN177 treatment or knockout of QPCTL also promoted the phagocytosis of A431 cells. Thus, QPCTL inhibitors increased the killing of tumor cells by both human neutrophils and macrophages by interrupting the CD47-SIRPα axis (Logtenberg et al. 2019). Regarding safety, the result of the first-in-man oral dose study that evaluated the safety and pharmacokinetics of PQ912 in healthy subjects showed that PQ912 was safe and well-tolerated. The results of a phase IIa study also showed that no significant difference existed between PQ912 (800 mg twice daily) and placebo in terms of the number of subjects with adverse events. Notably, no obvious hematological toxicity was observed in subjects treated with PQ912 (Scheltens et al. 2018). Consistent with this, QPCTL knockout mice were also viable and healthy (Becker et al. 2016). However, the mechanism was not clear thus far. QPCTL mediated CD47 protein modification at the early stage of the protein life cycle. Post-translational modification of QPCTL cannot be conducted in mature RBCs that lack a nucleus and organelles, thus, these RBCs may be less affected by QPCTL inhibitors. Furthermore, given that QPCTL is an enzyme, QPCTL inhibitors would not suffer from the “antigen sink” problem of anti-CD47 therapy (Ingram et al. 2017). Finally, small molecule inhibitors of QPCTL have advantages in terms of delivery and bioavailability. Moreover, they don’t cause hemagglutination easily. However, due to nonspecific modification inhibition for CD47, whether QPCTL inhibitors have other off-target effects such as the inhibition of glutaminyl-peptide cyclotransferase (QPCT) should be further explored.
Discussion and conclusions
The five strategies that diminish CD47 blockade-induced hematotoxicity are mechanically different and possess specific advantages compared with others. From the aspect of efficacy, bispecific and discriminating antibodies are more promising. The activation of pro-phagocytic signals is critical for CD47 blocking therapy (Chen et al. 2017); bispecific and discriminating antibodies can be formed with Fc fragments that potently activate pro-phagocytic signals, thereby generating single drugs that more effectively promote phagocytosis and kill tumor cells (Veillette and Chen 2018). These antibodies can more strongly promote innate immunity and enhance antigen presentation; they also exhibit better efficacy when combined with PD-1/PD-L1 antibodies that activate adaptive immunity, thereby improving tumor treatment (Feng et al. 2019).
In addition to the above-mentioned five strategies used to mitigate the hematotoxicity of CD47 blockade, some other notable approaches have the ability or potential to reduce the toxicity of CD47 blockade therapy, as follows. (1) ALX148, an engineered SIRPα fusion protein with high affinity yet insufficient pro-phagocytosis ability for tumor cells and erythrocytes, is used to block CD47. With the combination treatment of ALX148 and anti-TAA antibody such as anti-HER2 trastuzumab or anti-CD20 obinutuzumab, which could promote phagocytosis of tumor cells, the tumor cell was effectively killed, whereas few erythrocytes were eliminated (Kauder et al. 2018). (2) RRx-001 is a small molecule compound that reduces the CD47 and SIRPα expression. RRx-001 also did not cause hematotoxicity in clinical trials (Oronsky et al. 2021; Yu et al. 2020). (3) Pep-20, a peptide specifically targeting CD47, reportedly has no significant effect on RBCs in preclinical experiments (Wang et al. 2020b). (4) Delivery by nanoparticles targeting the tumor microenvironment could reduce CD47 antibodies’ contact with erythrocytes and can reduce hematotoxicity (Chen et al. 2019). (5) Antisense therapy targeting CD47 expression is also a promising strategy to decrease hematotoxicity due to the lack of CD47 transcription in mature erythrocytes (Schwartz et al. 2019).
Testing whether the CD47 blocking therapy significantly affects the human erythrocytes and platelets at the beginning is preferable due to the unneglectable hematotoxicity of CD47 antibodies. For example, in the development of CD47 antibody TJC4, the researchers excluded the CD47 antibodies that are highly bound to erythrocytes at an early step (Meng et al. 2019). Early detection of the potential toxicity during antibody development can help improve the drug development success rate. Performing safety assessment in animal models is needed. However, the use of animal models for the safety evaluation of CD47 blockade still has some limitations. The difference in the binding of the SIRPα fusion protein TTI-621 to human and cynomolgus monkey erythrocytes (Petrova et al. 2017) suggested that the safety evaluation of CD47 blockade in non-human primate experiments may not correspond to that in humans. In addition, to evaluate CD47 blocking, the affinity of nude mouse SIRPα to human CD47 was 65 times that of other mice strains (Kwong et al. 2014).
Considering the differences of CD47 and SIRPα between human and animal models, using humanized CD47 and SIRPα transgenic animals for safety evaluation may be a feasible solution. Human SIRPα knock-in xenograft mouse models have been used to study human tumors to better mimic the human CD47-SIRPα axis (Jinnouchi et al. 2020). It may also be used for safety evaluation in future studies. However, transgenic animals still cannot completely imitate human CD47 and SIRPα due to post-translational modifications and different interacting proteins. As a compromise method, safety evaluation is performed from multiple aspects to improve the safety of the CD47 blockade strategy.
In addition, the decrease of lymphocyte including T cells is another targeting CD47-related adverse event, which is neglected in the safety evaluation of CD47 blocking therapy development. Sikic et al. (2019) reported that 33% of the lymphocyte count decrease occurred in patients treated with Hu5F9-G4 alone. Pettersen et al. (1999) demonstrated that under appropriate conditions, CD47 activation resulted in very rapid T cell death. Considering the critical role of T cells in antitumor immunity and the promising future of the combination of CD47 block therapy and T cell immune checkpoint inhibitors, the evaluation of T cell toxicity should be paid more attention. CD47 was also expressed on platelets and hematopoietic stem cells (Jaiswal et al. 2009; Olsson et al. 2005). Thrombocytopenia was observed in the clinical research results of TTI-621, but it was transient and reversible, and was not associated with bleeding (Ansell et al. 2021). As for hematopoietic stem cells, CD47 antibodies enabled phagocytosis of leukemia stem cells by macrophages while sparing normal hematopoietic stem cells in vitro (Majeti et al. 2009). Therefore, the toxicity of platelets and hematopoietic stem cells may not be classified as serious side effects of CD47 blockade agents.
In conclusion, CD47 as a phagocytosis checkpoint is a promising target for tumor therapy. Although earlier anti-CD47 antibody treatment might induce hematotoxicity, multiple strategies have been proposed to diminish such toxicity. Therefore, we demonstrated the important role of hematotoxicity-free CD47 blockade in tumor treatment.
Data availability
Not applicable.
Abbreviations
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- ADCP:
-
Antibody-dependent cellular phagocytosis
- CD19:
-
Cluster of differentiation 19
- CD20:
-
Cluster of differentiation 20
- CD47:
-
Cluster of differentiation 47
- CTLA-4:
-
Cytotoxic T lymphocyte-associated molecule-4
- DLBCL:
-
Diffuse large B-cell lymphoma
- EGFR:
-
Epidermal growth factor receptor
- HER2:
-
Human epidermal growth factor receptor-2
- MTD:
-
Maximum tolerated dose
- PD-1:
-
Programmed cell death receptor-1
- PD-L1:
-
Programmed cell death ligand-1
- QPCT:
-
Glutaminyl-peptide cyclotransferase
- QPCTL:
-
Glutaminyl-peptide cyclotransferase like
- RBC:
-
Red blood cell
- SIRPα:
-
Signal regulatory protein alpha
- SIRPβ:
-
Signal regulatory protein beta
- SIRPγ:
-
Signal regulatory protein gamma
- TAA:
-
Tumor-associated antigens
References
Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N et al (2018) CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N Engl J Med 379(18):1711–1721
Agoram B, Wang B, Sikic BI, Lakhani NJ, Patnaik A, Liu J et al (2018) Pharmacokinetics of Hu5F9-G4, a first-in-class anti-CD47 antibody, in patients with solid tumors and lymphomas. J Clin Oncol 36(15_suppl):2525
Ansell S, Chen RW, Flinn IW, Maris MB, O’Connor OA, Johnson LD et al (2016) A phase 1 study of TTI-621, a novel immune checkpoint inhibitor targeting CD47, in patients with relapsed or refractory hematologic malignancies. Blood 128(22):1812
Ansell SM, Flinn IW, Maris MB, O’Connor OA, Lesokhin A, Advani AS et al (2017) TTI-621 (SIRPαFc), an immune checkpoint inhibitor blocking the CD47 “do not eat” signal, induces objective responses in patients with advanced, relapsed/refractory diffuse large B-cell lymphoma (DLBCL). Blood 130(Supplement 1):4116
Ansell SM, Maris MB, Lesokhin AM, Chen RW, Flinn IW, Sawas A et al (2021) Phase I study of the CD47 blocker TTI-621 in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res 27(8):2190–2199
Arias CF, Arias CF (2017) How do red blood cells know when to die? R Soc Open Sci 4(4):160850
Barazi HO, Li Z, Cashel JA, Krutzsch HC, Annis DS, Mosher DF et al (2002) Regulation of integrin function by CD47 ligands. Differential effects on alpha vbeta 3 and alpha 4beta1 integrin-mediated adhesion. J Biol Chem 277(45):42859–42866
Becker A, Eichentopf R, Sedlmeier R, Waniek A, Cynis H, Koch B et al (2016) IsoQC (QPCTL) knock-out mice suggest differential substrate conversion by glutaminyl cyclase isoenzymes. Biol Chem 397(1):45–55
Bouchlaka MN, Puro R, Capoccia B, Donio M, Hiebsch R, Carter AJ et al (2018) Development of AO-176, a next generation humanized anti-CD47 antibody with novel anti-cancer properties and negligible binding to red blood cells. Eur J Cancer 103:76
Bruce LJ, Ghosh S, King MJ, Layton DM, Mawby WJ, Stewart GW et al (2002) Absence of CD47 in protein 4.2-deficient hereditary spherocytosis in man: an interaction between the Rh complex and the band 3 complex. Blood 100(5):1878–1885
Buatois V, Johnson Z, Salgado-Pires S, Papaioannou A, Hatterer E, Chauchet X et al (2018) Preclinical development of a bispecific antibody that safely and effectively targets CD19 and CD47 for the treatment of B-Cell lymphoma and leukemia. Mol Cancer Ther 17(8):1739–1751
Chen J, Zhong MC, Guo H, Davidson D, Mishel S, Lu Y et al (2017) SLAMF7 is critical for phagocytosis of haematopoietic tumour cells via Mac-1 integrin. Nature 544(7651):493–497
Chen JY, McKenna KM, Choi TS, Duan J, Brown L, Stewart JJ et al (2018) RBC-specific CD47 pruning confers protection and underlies the transient anemia in patients treated with anti-CD47 antibody 5F9. Blood 132(Supplement 1):2327
Chen Q, Wang C, Zhang X, Chen G, Hu Q, Li H et al (2019) In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat Nanotechnol 14(1):89–97
Dheilly E, Moine V, Broyer L, Salgado-Pires S, Johnson Z, Papaioannou A et al (2017) Selective blockade of the ubiquitous checkpoint receptor CD47 Is enabled by dual-targeting bispecific antibodies. Mol Ther 25(2):523–533
Dheilly E, Majocchi S, Moine V, Didelot G, Broyer L, Calloud S et al (2018) Tumor-directed blockade of CD47 with bispecific antibodies induces adaptive antitumor immunity. Antibodies (basel) 7(1):3
Durand J, Gauttier V, Morello A, Pengam S, Vanhove B, Poirier N (2018) Abstract 1753: SIRPa inhibition monotherapy leads to dramatic change in solid tumor microenvironment and prevents metastasis development. Cancer Res 78(13 Supplement):1753
Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL (2019) Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer 19(10):568–586
Fernandes HP, Cesar CL, Barjas-Castro ML (2011) Electrical properties of the red blood cell membrane and immunohematological investigation. Rev Bras Hematol Hemoter 33(4):297–301
Freeman SA, Grinstein S (2014) Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol Rev 262(1):193–215
Gauttier V, Pengam S, Durand J, Morello A, Conchon S, Vanhove B et al (2018) Abstract 1684: Selective SIRPa blockade potentiates dendritic cell antigen cross-presentation and triggers memory T-cell antitumor responses. Cancer Res 78(13 Supplement):1684
Gauttier V, Pengam S, Durand J, Biteau K, Mary C, Morello A et al (2020) Selective SIRPalpha blockade reverses tumor T cell exclusion and overcomes cancer immunotherapy resistance. J Clin Invest 130(11):6109–6123
Ingram JR, Blomberg OS, Sockolosky JT, Ali L, Schmidt FI, Pishesha N et al (2017) Localized CD47 blockade enhances immunotherapy for murine melanoma. Proc Natl Acad Sci USA 114(38):10184–10189
Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R et al (2009) CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138(2):271–285
Jinnouchi F, Yamauchi T, Yurino A, Nunomura T, Nakano M, Iwamoto C et al (2020) Establishment of a human SIRPA knock-in xenograft mouse model to study human hematopoietic and cancer stem cells. Blood 135(19):1661–1672
Johnson LDS, Banerjee S, Kruglov O, Viller NN, Horwitz SM, Lesokhin A et al (2019) Targeting CD47 in Sezary syndrome with SIRPalphaFc. Blood Adv 3(7):1145–1153
Katz BZ, Herishanu Y (2014) Therapeutic targeting of CD19 in hematological malignancies: past, present, future and beyond. Leuk Lymphoma 55(5):999–1006
Kauder SE, Kuo TC, Harrabi O, Chen A, Sangalang E, Doyle L et al (2018) ALX148 blocks CD47 and enhances innate and adaptive antitumor immunity with a favorable safety profile. PLoS ONE 13(8):e0201832
Kaur S, Kuznetsova SA, Pendrak ML, Sipes JM, Romeo MJ, Li Z et al (2011) Heparan sulfate modification of the transmembrane receptor CD47 is necessary for inhibition of T cell receptor signaling by thrombospondin-1. J Biol Chem 286(17):14991–15002
Killian ML (2014) Hemagglutination assay for influenza virus. Methods Mol Biol 1161:3–9
Kwong LS, Brown MH, Barclay AN, Hatherley D (2014) Signal-regulatory protein alpha from the NOD mouse binds human CD47 with an exceptionally high affinity—implications for engraftment of human cells. Immunology 143(1):61–67
Lindberg FP, Gresham HD, Schwarz E, Brown EJ (1993) Molecular cloning of integrin-associated protein: an immunoglobulin family member with multiple membrane-spanning domains implicated in alpha v beta 3-dependent ligand binding. J Cell Biol 123(2):485–496
Liu J, Wang L, Zhao F, Tseng S, Narayanan C, Shura L et al (2015) Pre-clinical development of a humanized Anti-CD47 antibody with anti-cancer therapeutic potential. PLoS ONE 10(9):e0137345
Liu B, Guo H, Xu J, Qin T, Guo Q, Gu N et al (2018a) Elimination of tumor by CD47/PD-L1 dual-targeting fusion protein that engages innate and adaptive immune responses. Mabs 10(2):315–324
Liu X, Liu L, Ren Z, Yang K, Xu H, Luan Y et al (2018b) Dual targeting of innate and adaptive checkpoints on tumor cells limits immune evasion. Cell Rep 24(8):2101–2111
Liu Y, Chang Y, He X, Cai Y, Jiang H, Jia R et al (2020) CD47 enhances cell viability and migration ability but inhibits apoptosis in endometrial carcinoma cells via the PI3K/Akt/mTOR signaling pathway. Front Oncol 10:1525
Logtenberg MEW, Jansen JHM, Raaben M, Toebes M, Franke K, Brandsma AM et al (2019) Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPalpha axis and a target for cancer immunotherapy. Nat Med 25(4):612–619
Ma L, Zhu M, Gai J, Li G, Chang Q, Qiao P et al (2020) Preclinical development of a novel CD47 nanobody with less toxicity and enhanced anti-cancer therapeutic potential. J Nanobiotechnology 18(1):12
Mair B, Aldridge PM, Atwal RS, Philpott D, Zhang M, Masud SN et al (2019) High-throughput genome-wide phenotypic screening via immunomagnetic cell sorting. Nat Biomed Eng 3(10):796–805
Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr et al (2009) CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138(2):286–299
Matlung HL, Babes L, Zhao XW, van Houdt M, Treffers LW, van Rees DJ et al (2018) Neutrophils kill antibody-opsonized cancer cells by trogoptosis. Cell Rep 23(13):3946–59 e6
McCracken MN, Cha AC, Weissman IL (2015) Molecular pathways: activating T cells after cancer cell phagocytosis from blockade of CD47 “Don’t Eat Me” signals. Clin Cancer Res 21(16):3597–3601
Meng Z, Wang Z, Guo B, Cao W, Shen H (2019) TJC4, a differentiated anti-CD47 antibody with novel epitope and RBC sparing properties. Blood 134(Supplement_1):4063
Mohammed R, Milne A, Kayani K, Ojha U (2019) How the discovery of rituximab impacted the treatment of B-cell non-Hodgkin’s lymphomas. J Blood Med 10:71–84
Mouro-Chanteloup I, Delaunay J, Gane P, Nicolas V, Johansen M, Brown EJ et al (2003) Evidence that the red cell skeleton protein 4.2 interacts with the Rh membrane complex member CD47. Blood 101(1):338–344
Muramatsu M, Gonzalez HD, Cacciola R, Aikawa A, Yaqoob MM, Puliatti C (2014) ABO incompatible renal transplants: good or bad? World J Transplant 4(1):18–29
Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP (2000) Role of CD47 as a marker of self on red blood cells. Science 288(5473):2051–2054
Olsson M, Oldenborg PA (2008) CD47 on experimentally senescent murine RBCs inhibits phagocytosis following Fcgamma receptor-mediated but not scavenger receptor-mediated recognition by macrophages. Blood 112(10):4259–4267
Olsson M, Bruhns P, Frazier WA, Ravetch JV, Oldenborg PA (2005) Platelet homeostasis is regulated by platelet expression of CD47 under normal conditions and in passive immune thrombocytopenia. Blood 105(9):3577–3582
Oronsky B, Cabrales P, Caroen S, Guo X, Scribner C, Oronsky A et al (2021) RRx-001, a downregulator of the CD47- SIRPalpha checkpoint pathway, does not cause anemia or thrombocytopenia. Expert Opin Drug Metab Toxicol 17(4):355–357
Petrova PS, Viller NN, Wong M, Pang X, Lin GH, Dodge K et al (2017) TTI-621 (SIRPalphaFc): a CD47-blocking innate immune checkpoint inhibitor with broad antitumor activity and minimal erythrocyte binding. Clin Cancer Res 23(4):1068–1079
Pettersen RD, Hestdal K, Olafsen MK, Lie SO, Lindberg FP (1999) CD47 signals T cell death. J Immunol 162(12):7031–7040
Piccione EC, Juarez S, Liu J, Tseng S, Ryan CE, Narayanan C et al (2015) A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. Mabs 7(5):946–956
Piccione EC, Juarez S, Tseng S, Liu J, Stafford M, Narayanan C et al (2016) SIRPalpha-antibody fusion proteins selectively bind and eliminate dual antigen-expressing tumor cells. Clin Cancer Res 22(20):5109–5119
Puro RJ, Bouchlaka MN, Hiebsch RR, Capoccia BJ, Donio MJ, Manning PT et al (2020) Development of AO-176, a next-generation humanized anti-CD47 antibody with novel anticancer properties and negligible red blood cell binding. Mol Cancer Ther 19(3):835–846
Ribas A, Wolchok JD (2018) Cancer immunotherapy using checkpoint blockade. Science 359(6382):1350–1355
Ring NG, Herndler-Brandstetter D, Weiskopf K, Shan L, Volkmer JP, George BM et al (2017) Anti-SIRPalpha antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc Natl Acad Sci USA 114(49):E10578–E10585
Rizzo C, Caruso C, Vasto S (2014) Possible role of ABO system in age-related diseases and longevity: a narrative review. Immun Ageing 11:16
Sallman DA, Asch AS, Al Malki MM, Lee DJ, Donnellan WB, Marcucci G et al (2019) The first-in-class anti-CD47 antibody magrolimab (5F9) in combination with azacitidine is effective in MDS and AML patients: ongoing phase 1b results. American Society of Hematology, Washington
Sallman DA, Malki MA, Asch AS, Lee DJ, Kambhampati S, Donnellan WB et al (2020) Tolerability and efficacy of the first-in-class anti-CD47 antibody magrolimab combined with azacitidine in MDS and AML patients: phase Ib results. J Clin Oncol 38(15_suppl):7507
Scheltens P, Hallikainen M, Grimmer T, Duning T, Gouw AA, Teunissen CE et al (2018) Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer’s disease: results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimers Res Ther 10(1):107
Schwartz AL, Nath PR, Allgauer M, Lessey-Morillon EC, Sipes JM, Ridnour LA et al (2019) Antisense targeting of CD47 enhances human cytotoxic T-cell activity and increases survival of mice bearing B16 melanoma when combined with anti-CTLA4 and tumor irradiation. Cancer Immunol Immunother 68(11):1805–1817
Sharma P, Allison JP (2015) The future of immune checkpoint therapy. Science 348(6230):56–61
Shi R, Chai Y, Duan X, Bi X, Huang Q, Wang Q et al (2020) The identification of a CD47-blocking “hotspot” and design of a CD47/PD-L1 dual-specific antibody with limited hemagglutination. Signal Transduct Target Ther 5:16
Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D et al (2019) First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J Clin Oncol 37(12):946–953
Sim J, Sockolosky JT, Sangalang E, Izquierdo S, Pedersen D, Harriman W et al (2019) Discovery of high affinity, pan-allelic, and pan-mammalian reactive antibodies against the myeloid checkpoint receptor SIRPalpha. Mabs 11(6):1036–1052
Subramanian S, Tsai R, Sen S, Dahl KN, Discher DE (2006) Membrane mobility and clustering of Integrin Associated Protein (IAP, CD47)–major differences between mouse and man and implications for signaling. Blood Cells Mol Dis 36(3):364–372
Veillette A, Chen J (2018) SIRPalpha-CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol 39(3):173–184
Velliquette RW, Aeschlimann J, Kirkegaard J, Shakarian G, Lomas-Francis C, Westhoff CM (2019) Monoclonal anti-CD47 interference in red cell and platelet testing. Transfusion 59(2):730–737
Voets E, Parade M, Lutje Hulsik D, Spijkers S, Janssen W, Rens J et al (2019) Functional characterization of the selective pan-allele anti-SIRPalpha antibody ADU-1805 that blocks the SIRPalpha-CD47 innate immune checkpoint. J Immunother Cancer 7(1):340
Wang Y, Ni H, Zhou S, He K, Gao Y, Wu W et al (2020a) Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol Immunother 70(2):365–376
Wang H, Sun Y, Zhou X, Chen C, Jiao L, Li W et al (2020b) CD47/SIRPalpha blocking peptide identification and synergistic effect with irradiation for cancer immunotherapy. J Immunother Cancer 8(2):e000905
Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS et al (2012) The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA 109(17):6662–6667
Wu Z, Weng L, Zhang T, Tian H, Fang L, Teng H et al (2019) Identification of Glutaminyl Cyclase isoenzyme isoQC as a regulator of SIRPalpha-CD47 axis. Cell Res 29(6):502–505
Yanagita T, Murata Y, Tanaka D, Motegi SI, Arai E, Daniwijaya EW et al (2017) Anti-SIRPalpha antibodies as a potential new tool for cancer immunotherapy. JCI Insight 2(1):e89140
Yang Y, Guo R, Chen Q, Liu Y, Zhang P, Zhang Z et al (2018) A novel bispecific antibody fusion protein co-targeting EGFR and CD47 with enhanced therapeutic index. Biotechnol Lett 40(5):789–795
Yu WB, Ye ZH, Chen X, Shi JJ, Lu JJ (2020) The development of small-molecule inhibitors targeting CD47. Drug Discov Today 26(2):561–568
Acknowledgements
We express great thanks to Dr. Le-Le Zhang for the language polishing to our manuscript.
Funding
This work was funded by The Science and Technology Development Fund, Macau SAR (File no. 0129/2019/A3) and was partially supported by National Natural Science Foundation of China (81973516).
Author information
Authors and Affiliations
Contributions
Y-CC and WS wrote the main manuscript text. Y-CC drew the figures. J-JS, Y-CC and WS collected and analyzed the data. J-JL and J-JS provided suggestions and revisions. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declared that there is no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors approved the final manuscript for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, YC., Shi, W., Shi, JJ. et al. Progress of CD47 immune checkpoint blockade agents in anticancer therapy: a hematotoxic perspective. J Cancer Res Clin Oncol 148, 1–14 (2022). https://doi.org/10.1007/s00432-021-03815-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-021-03815-z