
Abstract
Purpose
Molecular mechanisms of response to hypomethylating agents in patients with myelodysplastic syndromes (MDS) and chronic myelomonocytic leukemia (CMML) still remain largely unknown. Therefore, the effects of 5-Azacytidine (Aza) on clonal architecture and DNA methylation were investigated in this study.
Methods
Using next-generation sequencing (NGS), 30 myeloid leukemia-associated genes were analyzed in 15 MDS/CMML patients with excellent response to Aza. Effects on methylation levels were analyzed by quantitative methylation analysis using pyrosequencing for the global methylation marker LINE-1 in patients and myeloid cell lines. Various myeloid cell lines and a healthy cohort were screened for methylation levels in 23 genes. Selected targets were verified on the MDS/CMML cohort.
Results
The study presented here showed a stable variant allele frequency and stable global methylation levels in responding patients. A significant demethylation of EZH2 and NOTCH1 was revealed in patients with Aza response.
Conclusions
A response to Aza is not associated with eradication of malignant clones, but rather with a stabilization of the clonal architecture. We suggest changes in CpG methylation levels of EZH2 and NOTCH1 as potential targets of epigenetic response to Aza treatment which may also serve as useful biomarkers after clinical evaluation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
5-Azacytidine (Aza) is approved for clinical use in patients with myeloid malignancies not eligible for hematopoietic stem cell transplantation (European Medicines Agency 2018). It is beneficial to patients with myelodysplastic syndromes (MDS), chronic myelomonocytic leukemia (CMML) and acute myeloid leukemia (AML), significantly improving overall survival (Fenaux et al. 2009; Fenaux and Ades 2009; Ades et al. 2013; Fenaux et al. 2010; Pleyer and Greil 2015). A common effect of chemotherapeutic agents or targeted therapies used in the treatment of myeloid disorders is the eradication of malignant clones. Nowadays, measuring the mutation allele burden of malignant clones is widely used to monitor treatment response. Recently it was reported that the mutation allele burden of CMML patients responding to the hypomethylating agents Aza or Decitabine remained largely unchanged by therapy (Merlevede et al. 2016). This finding raised the question of what mechanisms are truly responsible for treatment response to hypomethylating agents, suggesting epigenetic mechanisms. Previous investigations suggested that low doses of Aza cause DNA demethylation by depletion of DNA methyltransferases in various AML cell lines (Hollenbach et al. 2010). In this context, we sought to investigate the influence of Aza on clonal architecture of patients, by assessing the kinetics of individual somatic mutations using next-generation sequencing. To assess the effect of Aza on DNA methylation, we focused on methylation changes in CpG islands (CGIs). CGIs are interspersed DNA sequences which are GC-rich, CpG-rich, (Deaton and Bird 2011) and overlap promoter regions of 60–70% of all human genes (Illingworth and Bird 2009). Next to transcription start sites, CGIs are also found in gene bodies and intergenic regions (Jones 2012). Approximately half of all CpGs are found within repetitive elements, including the promoter region of LINE-1 (Xie et al. 2009; Lander et al. 2001). LINE elements belong to the classes of retroelements called LINEs (long interspersed elements) (Lander et al. 2001). These elements comprise 10% of all 28 million CpG sites in the human genome (Zheng et al. 2017). Since methylation in repetitive elements has been shown to correlate with global genomic DNA methylation content because of their high occurrence throughout the genome, LINE-1 has been used as surrogate marker for estimating the genomic DNA methylation level (Barchitta et al. 2014). LINE-1 is usually heavily methylated (Yang et al. 2004), thus we hypothesized it should be suitable to depict changes in methylation patterns caused by hypomethylating agents. In addition to repetitive elements, methylation levels of 23 leukemia-associated target genes (Rinke et al. 2013; Schäfer et al. 2016) were investigated to find potential molecular mechanisms for treatment response. These genes are known to be involved in signaling pathways (FLT3, JAK2, KRAS, BRAF, CBL, KIT, SH2B3, NOTCH1, SETBP1), epigenetic regulation (EZH2, KDM6A, ASXL1, DNMT3A, TET2, IDH1, IDH2), the RNA-splicing machinery (SF3B1, ZRSR2), the Cohesin-complex (STAG2) and transcriptional regulation (NPM1, RUNX1, WT1, CALR).
Materials and methods
Patients and healthy controls
Out of more than 100 MDS and CMML patients treated with Aza, patients with excellent cytological response to the epigenetic treatment were selected for further investigation. Bone marrow aspirates from 15 patients with MDS (n = 10) or CMML (n = 5) were obtained at the time of initial diagnosis and best cytological response to Aza treatment. This study included 9 male and 6 female patients with a median age of 62 years (range 48–74 years). Aza was given for a median of five treatment cycles (range 2–11 cycles). The median treatment duration was 8 months. Total leukocytes were isolated after red cell lysis. Constitutional DNA from all patients was obtained from oral mucosa cells using buccal swabs. A detailed description of the patients’ diagnoses and response to the treatment can be found in the Supplemental Data Table S1. Informed consent was provided for all patients according to the Declaration of Helsinki. As a control, leukocytes from peripheral blood of ten healthy test persons (male, n = 4; female, n = 6; median age, 33 years; range 24–58 years) were isolated after red cell lysis.
Cell culture and treatment
Eleven human myeloid leukemia cell lines (HEL, ELF-153, HL-60, SKM-1, THP-1, MOLM-13, OCI-AML3, MV4-11, NB-4, K-562, BV-173) were cultivated in 80% RPMI 1640 medium with 20% FCS. The growth medium for ELF-153 was additionally supplemented with GM-CSF (5 ng/ml). ELF-153 and MOLM-13 were incubated for 72 h with their cell line-specific half maximal inhibitory concentration (IC50) of Aza (Sigma-Aldrich, St. Louis, MO, United States), applied as single dose. IC50 were determined using the MTT Assay (Promega, Fitchburg, WI, United States).
DNA isolation
Genomic DNA (gDNA) was extracted, using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. DNA of each patient was then amplified by whole-genome amplification (WGA), using the REPLI-g Ultra Fast Mini Kit (Qiagen) for further NGS analysis.
Next-generation sequencing
Next-generation sequencing (NGS) and data analysis were performed for 30 leukemia-associated target genes as previously described (Schäfer et al. 2016). Briefly, 231 amplicons and 8 external JAK2-V617F sensitivity controls were prepared for each patient sample and processed in a single NGS run. An independent NGS run was performed with original gDNA to confirm all mutations found in whole-genome amplified DNA. All mutational percentages listed refer to the results found in gDNA. The somatic origin was confirmed by NGS, using constitutional DNA obtained from buccal swabs.
Quantitative methylation analysis by pyrosequencing
Global methylation levels of patient samples and cell lines were determined using the surrogate marker LINE-1. LINE-1 primers were used as described by Bollati et al. (2007), covering three CpGs. Out of 30 genes investigated by NGS, CpG assays (PyroMark CpG Assays, Qiagen) were commercially available for 23 genes (ASXL1, DNMT3A, EZH2, IDH1, IDH2, KDM6A, TET2, STAG2, CALR, NPM1, RUNX1, WT1, SF3B1, ZRSR2, BRAF, CBL, FLT3, JAK2, KIT, KRAS, NOTCH1, SETBP1, SH2B3). These CpG assays mostly cover annotated promoter regions, but also intragenic CGIs. A list of CGI loci and their functions as listed in the Ensembl database is included in the Supplemental Data Table S2. Genomic DNA was treated with sodium bisulfite using the EpiTect Bisulfite Kit (Qiagen). Converted gDNA was amplified using the PyroMark PCR kit (Qiagen). Pyrosequencing analysis was conducted using 2 ng template material of myeloid cell lines and an increased amount of 25 ng for patients and incubated cells, respectively, to improve accuracy and reliability of the results (Supplemental Data Figure S1). A total of 269 CpG sites were investigated per sample. Peripheral blood leukocytes of ten healthy test persons were analyzed as controls. Methylation levels in ELF-153 and MOLM-13 were investigated after 24, 48, and 72 h of incubation with Aza (three biological replicates). Further analysis was conducted for 15 patients at initial diagnosis and best response to the treatment for verification of possible target genes of Aza. Primers and PCR conditions are listed in Supplemental Data Tables S2 and S3. PCR products were resolved on 3% agarose gels and visualized by staining with ethidium bromide, then sequenced on a PyroMark Q96 ID platform (Qiagen). Methylation levels for each CpG within the targeted region were quantified using the Pyro Q-CpG Software (Qiagen). For statistical analysis, data sets were tested for normality using the Shapiro–Wilk test. Comparisons of methylation levels were conducted using the Wilcoxon signed-rank test, provided by GraphPad Prism v6.01 (GraphPad Software, Inc., San Diego, CA, United States).
Results
Stable clonal architecture of Aza-responding patients as measured by targeted next generation sequencing
NGS was performed for 15 patients (MDS, n = 10; CMML, n = 5) at initial diagnosis and best cytological response to Aza treatment. A total of 47 somatic mutations were detected in 13 patients (in 8 of 10 MDS patients and, in 5 of 5 CMML patients) with some mutations appearing only at initial diagnosis, others only at best cytological response and 27 mutations appearing at both time points, thus allowing analysis of mutational kinetics (Fig. 1). A significant change in the mutational level was defined as a 25% difference in variant allele frequency between corresponding samples. In 70% of all mutations appearing at both time points, the variant allele frequency remained stable, suggesting a stable clonal architecture of patients responding to Aza treatment. Mutations appeared predominantly in epigenetic regulator genes, namely ASXL1, DNMT3A, EZH2, IDH2 and TET2 (Supplemental Data Figure S2). A list of all mutations found in the patient cohort is included in the Supplemental Data Table S4, cooperation of somatic mutations can be found in Supplemental Data Figure S3.

Stable clonal architecture of patients responding to Aza treatment. NGS was performed for 15 patients. Variant allele frequency remained stable in 70% of mutations appearing at both investigated time points
Stable global methylation levels of Aza-responding patients based on repetitive elements
LINE-1 displayed non-significant demethylation in patients (n = 15) (Fig. 2). Median methylation before treatment was 81.5% [interquartile range (IQR) 78.48–83.29], median methylation at best cytological response was 80.3% [IQR 78.24–84.79] (n.s.). Thus, LINE-1 is considered to maintain stable methylation patterns under treatment with Aza.

Stable CpG methylation levels of global methylation marker LINE-1 in patients responding to Aza treatment. LINE-1 displays non-significant demethylation under treatment with Aza (n = 15)
Four main gene targets of Aza identified by methylation analysis: EZH2, NOTCH1, RUNX1 and WT1
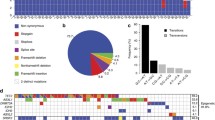
A systematic investigation of CpG methylation in myeloid cell lines was conducted. The complete screening can be found in Fig. 3. Moderate (20–80% methylation) up to high methylation levels (> 80% methylation) were observed in DNMT3A, EZH2, IDH2, TET2, RUNX1, WT1, FLT3, KIT, KRAS, NOTCH1, SETBP1 and SH2B3. Two genes, namely WT1 and NOTCH1, displayed hypermethylation in all 11 examined cell lines with an average methylation of > 80% per assay. In the healthy control group, only CpGs of EZH2, NOTCH1, RUNX1 and WT1 and were methylated > 20% (Fig. 3). Preliminary methylation analysis of genes involved in epigenetic regulation showed that a minimum methylation of 20% is necessary to be able to effectively measure demethylation in patients. As pre-analysis revealed matching patterns between the healthy control group and a subset of patients (Supplemental Figure S4a, S4b), we selected these four genes (EZH2, NOTCH1, RUNX1, WT1) for further investigation as possible target genes of Aza.

Characterization of CpG methylation in myeloid cell lines (n = 11) and a healthy control group (n = 10). Healthy test persons are labelled as #1–#10. A total of 269 CpGs from 23 genes were analyzed per sample. In the cell lines moderate (20–80%) up to high (> 80%) methylation levels were observed in CpGs of DNMT3A, EZH2, IDH2, TET2, RUNX1, WT1, FLT3, KIT, KRAS, NOTCH1, SETBP1 and SH2B3. EZH2, RUNX1, WT1 and NOTCH1 were also methylated in the healthy control group
Aza-caused demethylation in methylated CpG sites of ELF-153 and MOLM-13 cell lines
An in vitro evaluation of response to Aza was carried out for the cell lines ELF-153 and MOLM-13. The IC50 of Aza after 72 h was determined at 5.8 µmol/l for ELF-153 and at 2.5 µmol/l for MOLM-13 (Supplemental Data Figure S5). LINE-1 as marker of global methylation and possible target genes identified in the cell line screening (EZH2, NOTCH1, RUNX1 and WT1) were analyzed. Comparison of the untreated controls (at 0 h) with samples after 72 h of incubation revealed significant demethylation of LINE-1 and of all investigated genes (Fig. 4). Genes with a high baseline methylation (e.g. EZH2) showed larger absolute reduction in methylation than genes with a low baseline methylation (e.g. RUNX1). The largest reduction in methylation occurred in EZH2 with 46.1% in ELF-153 and 37.5% in MOLM-13. Information about the number of analyzed CpGs, median methylation levels of untreated controls and after 72 h of incubation with Aza are given in Supplement Data Table S5.

Decrease in CpG methylation levels of LINE-1, EZH2, NOTCH1, RUNX1 and WT1 in MOLM-13 and ELF-153 incubated with Aza for 72 h (three biological replicates each for MOLM-13 and ELF-153; LINE-1: n = 3 CpGs, EZH2: n = 3 CpGs, NOTCH1: n = 4 CpGs, RUNX1: n = 6 CpGs, WT1: n = 3 CpGs). Statistically significant decrease in methylation levels occurred over the course of 72 h in all investigated genes; **p ≤ 0.01, ***p ≤ 0.001
Verification of two gene targets in Aza-responding patients: EZH2 and NOTCH1
The four possible targets EZH2, NOTCH1, RUNX1 and WT1 were analyzed in patients (assays as listed in Supplemental Data Table S2: EZH2_1, NOTCH1_6, RUNX1_4, WT1_3). All tested CpG sites of two genes (EZH2 and NOTCH1) displayed significant demethylation at best cytological response of patients to the treatment with Aza. Exact methylation levels for specific CpGs are displayed in Fig. 5. Mapping and functions of targeted regions as annotated in the Ensembl database (release 91-December 2017) (Zerbino et al. 2018) are listed in Table 1. Methylation analyses for each assay showed the following results: median methylation of EZH2 (three CpGs) was reduced from 52.5 to 41%. Median methylation of NOTCH1 (four CpGs) was reduced from 84.4 to 70.1%. RUNX1 displayed non-significant demethylation in five out of six tested CpG sites, thus not qualifying as target. WT1 methylation showed a trend to demethylation, though not statistically significant (reduction from a median methylation of 41.0 to 28.1% for a total of three CpGs). Median methylation levels of verified targets for individual CpGs at initial diagnosis and at best cytological response with corresponding significance levels determined by Wilcoxon signed-rank test are given in Table 2.

Demethylation of gene-specific CpG sites of EZH2 and NOTCH1 in patients responding to Aza treatment. CpG Methylation analysis was conducted for the selected targets EZH2, NOTCH1, RUNX1 and WT1 in patients. Gene-specific CpG sites of EZH2 (promoter region) and NOTCH1 (intragenic region) show significant demethylation at the time of best cytological response to Aza treatment. WT1 shows a trend to demethylation (n = 15); *p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.0001
Discussion
In this study, effects of Aza therapy on clonal architecture and methylation were investigated. According to NGS data, a stable clonal architecture of MDS and CMML patients responding to Aza treatment was observed. Thus, a cytological response seems not associated with a reduction of the mutated clone, but more probable with a change in epigenetic features. Similar results have been presented before for CMML patients treated with hypomethylating agents. Merlevede et al. (2016) found that hypomethylating agents do not reduce mutation allele burden, as the size of the mutated clone remained unchanged in responding patients.
LINE-1 was analyzed in the patient cohort to gain information about global methylation levels. Surprisingly, in contrast to cell lines incubated with Aza, no difference in global methylation was detected in paired patients’ samples (initial diagnosis compared to best cytological response). This study suggests that a response to Aza is associated with a stable level of global methylation. However, temporary demethylation of LINE-1 may occur. In 2008, Stresemann et al. (2008) showed examples of five patients treated with Aza with transient reduction of global methylation levels within treatment cycles measured by capillary electrophoresis examined in peripheral blood mononuclear cells. However, methylation levels were not assessed at best response to the treatment as in the study presented here. Thus, stabilized global methylation levels may be a hallmark of well-responding patients.
To assess potential targets of epigenetic response to Aza treatment, a systematic characterization of CpG methylation in various myeloid cell lines and a healthy control group was conducted. Genes hypermethylated in cell lines and the healthy control group (EZH2, NOTCH1, RUNX1 and WT1) were selected as possible target genes of Aza. Elevated methylation levels of STAG2 in the healthy control group were regarded to be sex related, with gender being one of the accepted confounders in DNA methylation analyses (next to tissue type, cell type, age and smoking) (Chang et al. 2016; Zeilinger et al. 2013).
In vitro methylation levels were assessed in the cell lines ELF-153 and MOLM-13 over the course of 72 h after incubation with Aza. Quantitative methylation analyses were performed for LINE-1 and selected target genes. Methylation levels of all target genes were found to be decreasing over 72 h. The largest absolute reduction in methylation levels occurred in EZH2. This experiment indicated that looking for methylated genes seems an eligible step to find potential targets that could be used as biomarkers, since a response to Aza is measurable.
Finally, target genes were verified in patients. A significant demethylation of specific CpG sites of EZH2 and NOTCH1 was revealed in patients with excellent response to Aza treatment. Demethylation affected both promoter CGIs (EZH2) and intragenic CGIs (NOTCH1). Methylation of promoter regions has been largely accepted to down-regulate gene expression. A recent study challenges this generally accepted assumption, proposing that methylation affects the binding affinity of transcription factors to DNA, resulting in either positive or negative regulation of target genes (Ma et al. 2013). Ma et al. (2013) also found that, in some genes, promoter methylation did not affect gene expression at all. The function of gene body methylation remains unclear. Lately, a positive correlation has been reported between gene expression and gene body methylation (Jjingo et al. 2012) hypothesized before by Jones (1999). Maunakea et al. proposed that intragenic methylation might control alternative promoter usage, since most genes have at least two transcriptional start sites (one at the upstream promoter, another downstream within the gene body) (Jones 2012; Maunakea et al. 2010). Despite the functional role of CGI methylation still being discussed, DNA methylation remains a measurable quantitative epigenetic mark. Thus, measuring CpG methylation by pyrosequencing can be a useful method to reveal targets which may serve as biomarkers in future.
In addition to regulatory functions annotated in the Ensembl database, targeted regions were matched with histone modifications (Table 1) retrieved from the ENCODE project of K562 as representative for stem cell precursors of the myeloid lineage (Wang et al. 2013). Common histone modifications determined by ChiP-seq assay are H3K4me3 as a mark often found near promoters and H3K4me1 and H3K27ac often found near regulatory elements, e.g. enhancers (Calo and Wysocka 2013). The targeted regions of EZH2 span a promoter region marked by H3K4me3 located in the 5′ upstream region, and a transcriptionally active site marked by H3K4me1 and H3K27ac. The targeted regions of WT1 and RUNX1 are associated with distal enhancers due to the H3K4me1 mark and location within the gene body. The targeted region in NOTCH1 has not been associated with a regulatory genomic region. Analysis of identified candidate epigenetic markers in a prospective clinical trial will reveal whether they could serve as predictive markers, by correlating response or relapse to methylation levels, which has been previously discussed by Laird (2003).
In conclusion, at the genetic level somatic mutation analysis by NGS in responders revealed that a cytological response to Aza was not associated with a reduction of mutated clones, but rather with a stabilization of clonal architecture. At the epigenetic level, there were stable global methylation levels of Aza-responders on the one hand and measurable demethylation at gene-specific CpG sites on the other hand. This study shows that Aza modulates CpG methylation levels of EZH2 and NOTCH1 in MDS and CMML. This modulation affects either the promoter or the gene body of target genes including coding and non-coding regions. To our knowledge this is the first study suggesting that CpG methylation levels of EZH2 and NOTCH1 are targets for epigenetic response to Aza treatment.
References
Ades L, Sekeres MA, Wolfromm A, Teichman ML, Tiu RV, Itzykson R et al (2013) Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk Res 37(6):609–613
Barchitta M, Quattrocchi A, Maugeri A, Vinciguerra M, Agodi A (2014) LINE-1 hypomethylation in blood and tissue samples as an epigenetic marker for cancer risk: a systematic review and meta-analysis. PLoS One 9(10):e109478
Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D et al (2007) Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res 67(3):876–880
Calo E, Wysocka J (2013) Modification of enhancer chromatin: what, how, and why? Mol Cell 49(5):825–837
Chang CW, Lu TP, She CX, Feng YC, Hsiao CK (2016) Gene-set analysis with CGI information for differential DNA methylation profiling. Sci Rep 19(6):24666
Deaton AM, Bird A (2011) CpG islands and the regulation of transcription. Genes Dev 25(10):1010–1022
Fenaux P, Ades L (2009) Review of azacitidine trials in intermediate-2- and high-risk myelodysplastic syndromes. Leuk Res 33(Suppl 2):S7–11
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A et al (2009) Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 10(3):223–232
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Gattermann N, Germing U et al (2010) Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol 28(4):562–569
European Medicines Agency (2018) EMA/450923/2016 – Vidaza, Annex I, summary of product characteristics. https://www.ema.europa.eu/en/medicines/human/EPAR/vidaza
Hollenbach PW, Nguyen AN, Brady H, Williams M, Ning Y, Richard N et al (2010) A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS One 5(2):e9001
Illingworth RS, Bird AP (2009) CpG islands–‘a rough guide’. FEBS Lett 583(11):1713–1720
Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK (2012) On the presence and role of human gene-body DNA methylation. Oncotarget 3(4):462–474
Jones PA (1999) The DNA methylation paradox. Trends Genet 15(1):34–37
Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13(7):484–492
Laird PW (2003) The power and the promise of DNA methylation markers. Nat Rev Cancer 3(4):253–266
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J et al (2001) Initial sequencing and analysis of the human genome. Nature 409(6822):860–921
Ma X, Wang YW, Zhang MQ, Gazdar AF (2013) DNA methylation data analysis and its application to cancer research. Epigenomics 5(3):301–316
Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD et al (2010) Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466(7303):253–257
Merlevede J, Droin N, Qin T, Meldi K, Yoshida K, Morabito M et al (2016) Mutation allele burden remains unchanged in chronic myelomonocytic leukaemia responding to hypomethylating agents. Nat Commun 24(7):10767
Pleyer L, Greil R (2015) Digging deep into “dirty” drugs-modulation of the methylation machinery. Drug Metab Rev 47(2):252–279
Rinke J, Schafer V, Schmidt M, Ziermann J, Kohlmann A, Hochhaus A et al (2013) Genotyping of 25 leukemia-associated genes in a single work flow by next-generation sequencing technology with low amounts of input template DNA. Clin Chem 59(8):1238–1250
Schäfer V, Ernst J, Rinke J, Winkelmann N, Beck JF, Hochhaus A, Gruhn B, Ernst T (2016) EZH2 mutations and promoter hypermethylation in childhood acute lymphoblastic leukemia. J Cancer Res Clin Oncol 142(7):1641–1650
Stresemann C, Bokelmann I, Mahlknecht U, Lyko F (2008) Azacytidine causes complex DNA methylation responses in myeloid leukemia. Mol Cancer Ther 7(9):2998–3005
Wang H, Hu H, Zhang Q, Yang Y, Li Y, Hu Y et al (2013) Dynamic transcriptomes of human myeloid leukemia cells. Genomics 102(4):250–256
Xie H, Wang M, Bonaldo Mde F, Smith C, Rajaram V, Goldman S et al (2009) High-throughput sequence-based epigenomic analysis of Alu repeats in human cerebellum. Nucleic Acids Res 37(13):4331–4340
Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP (2004) A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res 32(3):e38
Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C et al (2013) Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One 8(5):e63812
Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J et al (2018) Ensembl 2018. Nucleic Acids Res 46(D1):D754–D761
Zheng Y, Joyce BT, Liu L, Zhang Z, Kibbe WA, Zhang W et al (2017) Prediction of genome-wide DNA methylation in repetitive elements. Nucleic Acids Res 45(15):8697–8711
Funding
This study was funded by the Interdisciplinary Center for Clinical Research (IZKF), Universitätsklinikum Jena, Germany.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gawlitza, A.L., Speith, J., Rinke, J. et al. 5-Azacytidine modulates CpG methylation levels of EZH2 and NOTCH1 in myelodysplastic syndromes. J Cancer Res Clin Oncol 145, 2835–2843 (2019). https://doi.org/10.1007/s00432-019-03016-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-019-03016-9