
Abstract
Background
Increasing evidence has shown that long non-coding RNAs (lncRNAs) are important in hepatocellular carcinoma (HCC) development and progression. In this study, we aim to evaluate the expression of lncRNA FAM99B and its biological function in HCC.
Methods
The expression level of FAM99B in HCC was assessed based on data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO), verified using quantitative real-time polymerase chain reaction (qRT-PCR). HCCLM3 was transfected with lentivirus containing full-length FAM99B to obtain stable overexpressing cell line. Cell Counting Kit 8, clone formation, and transwell assays were used to investigate the effects of FAM99B in HCC progression. In addition, Gene Ontology, Kyoto Encyclopedia of Genes and Genomes, and PANTHER pathway analyses were conducted to investigate the underlying molecular mechanisms.
Results
FAM99B was found to be downregulated in HCC tissues compared with adjacent normal tissues based on TCGA, GEO, and qRT-PCR data. Our results revealed that downregulated FAM99B was significantly associated with vascular invasion, advanced histologic grade, and T stage. Kaplan–Meier analysis using TCGA data indicated that decreased FAM99B levels were significantly associated with poor overall survival in patients with HCC. Moreover, overexpression of FAM99B significantly inhibited cell proliferation, migration, and invasion in vitro. Pathway analyses showed that the co-expressed genes of FAM99B mainly participated in the pathways “Metabolic pathways” and “Blood coagulation”.
Conclusion
Our results suggest that FAM99B may serve as a tumor suppressor in HCC and may provide a promising therapy target for patients with HCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Liver cancer is a major public health problem worldwide. It is estimated to be the sixth most prevalent malignancy and the fourth leading cause of cancer-related deaths globally, with an estimated 841,000 new cases and 782,000 mortalities in 2018 (Bray et al. 2018). Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer and accounts for approximately 80% of liver cancer cases. In the past decades, liver transplantation and hepatic resection have become the major treatments for HCC. Although great improvements have been made to the treatment of HCC, its recurrence rate remains high and a complete cure has yet to be found (Forner et al. 2018). Therefore, understanding the molecular mechanisms behind the development of HCC is essential for future diagnostic and therapeutic advances.
Recently, evidence has shown that long non-coding RNAs (lncRNAs), a classification of non-coding RNAs that are longer than 200 nucleotides, play an important role in HCC tumorigenesis and progression by interacting with DNA, proteins, and RNA (Lanzafame et al. 2018). Many studies have demonstrated that numerous dysregulated lncRNAs may contribute to the development and progression of HCC (He et al. 2014). Results from high-throughput RNA sequencing (RNA-Seq) of 12 paired HCC tissues and normal tissues have shown that multiple differentially expressed lncRNAs are notably associated with tumor cell differentiation, portal vein tumor thrombus, and AFP levels (Yao et al. 2017). Moreover, lncRNA miR503HG has been found to inhibit HCC tumor metastasis by regulating the NF-Κb-signaling pathway to exert its tumor suppression role (Wang et al. 2018). Another lncRNA, uc.134, has been shown to have low expression in HCC, which is significantly associated with the number of tumors, lymph node metastasis, TNM stage, and poor overall survival (OS) in patients with HCC (Ni et al. 2017). LncRNA 00607 has been identified as a novel tumor suppressor in HCC and its overexpression can inhibit HCC cell proliferation, promote cell apoptosis, and enhance the sensitivity of chemotherapeutic drugs (Sun et al. 2018). These findings suggest that novel aberrant lncRNAs may participate in the molecular mechanisms of HCC, contributing to its incidence and development. LncRNAs may also provide new targets for early diagnosis and treatment.
FAM99B, a family with sequence similarity 99-member B, is a novel antisense lncRNA that is 1066 bps in length and has no protein-coding function. A quantitative RNA-Seq of all the major human organs and tissues has revealed that FAM99B is only expressed in hepatic, placental, and testicular tissues, and is especially highly expressed in hepatic tissues (Fagerberg et al. 2014). As a representative liver-specific non-coding RNA, microRNA-122 has been suggested to be involved in HCC development and metastasis (Chang et al. 2004; Huang and He 2011). A novel, liver-specific lncRNA, LINC01093 has also been shown to have a tumor suppressive effect on HCC progression (He et al. 2019). FAM99B encoded by a gene located on chromosome 11p15.5 and loss of heterozygosity on chromosome 11p have been found to lead to an increased expression of a recessive oncogene in the genome and promote HCC development (Wang and Rogler 1988). Given these specificities, we hypothesized that FAM99B may be associated with HCC.
In the present study, we discovered that FAM99B may be involved in the tumorigenesis and progression of HCC. The expression of FAM99B is prominently decreased in HCC and its expression is inversely correlated with vascular invasion, lymph node metastasis, and tumor grade. Besides, patients with low FAM99B tend to have poor OS in HCC patients. Functional analyses indicated that the overexpression of FAM99B repress cell proliferation, migration, and invasion in vitro. As such, we first demonstrated that FAM99B may serve as a tumor suppressor in HCC.
Materials and methods
The expression level of FAM99B in HCC based on public online databases
RNA-Seq data of 371 HCC tissues and 50 adjacent normal tissues were extracted from The Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/) (up to October 30, 2018). The RNA expression data were represented as Transcripts per million (TPM) and normalized by converting to the base-2 logarithm. In addition, the relationships between FAM99B expression, clinicopathological features, as well as the prognosis were calculated.
We also retrieved the original lncRNA expression data from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) (up to October 19, 2018). The retrieval keywords were as follows: (1) lncRNAs, long non-coding RNAs, and non-coding RNAs; (2) cancer, tumor, carcinoma, neoplasm*, and malignant*; and (3) hepatocellular carcinoma, HCC, and liver. Data sets that met the following criteria were enrolled. (1) HCC tissues and adjacent normal liver tissues were included and each group contained more than three cases; (2) the expression data of FAM99B could be obtained or evaluated directly; and (3) the tissue samples were extracted from Homo sapiens. Two researchers (Meile Mo and Shun Liu) carefully screened for the eligible data sets and extracted basic information, including the GSE number, publication year, platform, country, sample size of each group, and the expression level of FAM99B. Finally, a meta-analysis was conducted using these GEO data sets.
Tissue samples
A total of 80 HCC tissues and their adjacent normal tissues (at least 3 cm away from HCC tissues) were obtained from patients who had undergone surgery in the Affiliated Cancer Hospital of Guangxi Medical University in China from March 2016 to September 2018. The tissue samples were stored in liquid nitrogen until use. All the patients with HCC were diagnosed by two experienced pathologists and had not received radiotherapy or chemotherapy treatment prior to surgery. All the participants signed informed consent forms before participating in the study. The research was approved by the Ethics Committee of Guangxi Medical University.
Cell culture and lentiviral infection
The human HCC cell lines MHCC97L, MHCC97H (CellCook Biotech Co. Ltd.), HCCLM3 (Zhongshan Hospital, Fudan University of Shanghai), and Huh-7, HepG2, Hep3B (Cell bank of the Chinese academy of sciences) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin and streptomycin (Gibco) in a humidified atmosphere at 37 °C containing 5% CO2.
The empty lentiviral vector (Lv-NC) and the GV502 lentiviral vector containing full-length human FAM99B (Lv-FAM99B) were constructed by Genechem (Shanghai, China). Lv-FAM99B and Lv-NC were introduced to HCCLM3 cells following the manufacturer’s instructions. After transfection, the cells were exposed to puromycin for 2 weeks to obtain clones with stable overexpression of FAM99B. The cells were harvested for RNA isolation and the infection efficiency was determined using qRT-PCR.
qRT-PCR
Total RNA was extracted from tissues and cells using a Trizol Reagent (Invitrogen) and then used to synthesize cDNA using High-Capacity cDNA Reverse Transcription Kits (Thermo Fisher Scientific) following the manufacturer’s instructions. Quantifications of FAM99B and GAPDH were conducted using the TB GreenTM Premix Ex TaqTM II Kit (Takara) in the StepOnePlus real-time PCR system (Applied Biosystems) following the manufacturer’s instructions. GAPDH was set as the internal control. The sequences for the GAPDH and FAM99B primers are as follows: GAPDH-F (5′–3′): AGCCACATCGCTCAGACAC, GAPDH-R (5′–3′): GCCCAATACGACCAAATCC; FAM99B-F (5′–3′): ACAGTGACCGCCGAGACA, FAM99B-R (5′–3′): AGATTTGGGATTTAGGGAAGG. The relative expression of FAM99B was calculated using the formula 2−△△Ct.
Cell proliferation, migration, and invasion assays
Cell proliferation viability was assessed using the CCK-8 assay (Dojindo) according to the manufacturer’s protocol. A total of 1000 cells per well were seeded into 96-well plates and incubated at different timepoints. 10 μl of CCK-8 was then added into each well and incubated for another 2 h, so that the absorbance value at 450 nm can be measured. To evaluate the cells’ cloning ability, cells were seeded into six-well plates (500 cells per well) and incubated for 2 weeks. After that, the colonies were fixed with methanol and stained with 0.1% crystal violet. Only colonies containing more than 50 cells were counted.
Transwell inserts (8 μm, Costar, Corning) coated with and without the Matrigel (Matrigel Basement Membrane Matrix, Corning) were placed into 24-well plates to measure cell invasion and migration ability, respectively. In the upper chambers, serum-free medium containing 1 × 105 cells per well were seeded for the invasion assay, and medium containing 5 × 104 cells per well were seeded for the migration assay. For the lower chambers, 600 μl of culture medium containing 20% FBS was added. After incubating for 48 h, cells remaining on the upper chambers were wiped off with cotton swaps, whereas cells that had migrated through/invaded the membrane were fixed with methanol and stained with 0.1% crystal violet. The number of migrated/invaded cells was counted using an inverted microscope with three randomly selected fields.
Prediction of the co-expressed genes of FAM99B and pathway analyses
The co-expressed genes of FAM99B were obtained from the following prediction websites: cBioPortal (Cerami et al. 2012; Gao et al. 2013) (http://www.cbioportal.org/), MEM (Adler et al. 2009) (https://biit.cs.ut.ee/mem/), and TANRIC databases (https://ibl.mdanderson.org/tanric/_design/basic/index.html) (Li et al. 2015). Intersected genes from at least two data sets were considered potentially related genes. Subsequently, KOBAS 3.0 (Xie et al. 2011) (http://kobas.cbi.pku.edu.cn/) was used to conduct GO enrichment, KEGG, and PANTHER pathway analyses. Corrected P values less than 0.05 were considered statistically significant.
Statistical analysis
Numerical variables are presented as mean ± standard deviation (SD). Student’s/paired t test was used to compare the difference between groups. The Chi-square test was used to assess the relationship between FAM99B expression level and clinicopathological features. The Kaplan–Meier plot and Log-rank test were conducted using a TCGA cohort (N = 370) to explore the survival significance of FAM99B in HCC. The receiver operating characteristic (ROC) curve was constructed to evaluate the diagnostic efficiency of FAM99B in HCC. The Youden index (sensitivity + specificity − 1) was used to set the optimal cutoff point. All analyses were conducted using SPSS 20.0, and the graphs were drawn using GraphPad Prism. P < 0.05 was defined as a statistically significant difference.
For the meta-analysis of GEO data sets, the standardized mean difference (SMD) with its 95% confidence interval (CI) was calculated using STATA 12.0. Considering the different experimental conditions and platforms among GEO data sets, a random-effect model was used to combine these results. Heterogeneity test was conducted using Chi-squared and I-squared tests to assess whether inter-study heterogeneity occurred, I2> 50% or P < 0.05 were considered statistically significant heterogeneity (DerSimonian and Laird 2015). A combined SMD < 0 with a 95% CI that did not include 0 suggested that FAM99B expression was significantly decreased in HCC tissues compared with normal tissues. Funnel plot and Begg’s test were used to assess the publication bias (Begg and Mazumdar 1994), and P > 0.1 indicated no obvious publication bias. Sensitivity analysis was performed to examine the stability of the meta-analysis by removing 1 study each time.
Results
FAM99B is downregulated in HCC tissues and correlated with poor survival based on TCGA database
Independent-sample t test showed that FAM99B expression was decreased in HCC tissues compared with noncancerous tissues (P < 0.001; Table 1; Fig. 1a). ROC curve analysis was conducted to determine the efficiency of FAM99B expression to distinguish HCC tissues from normal tissues. The area under the curve (AUC) of FAM99B in HCC of TCGA data was 0.828 (95% CI 0.788–0.862, P < 0.0001), and the cutoff value was 2.272. The sensitivity and specificity were 71.97% and 92.00%, respectively (Fig. 1b). The patients were divided into a high expression group (> 1.1774) and low expression group (≤ 1.1774) based on their median value of FAM99B expression. Then, we examined the relationship between FAM99B expression and clinicopathological features. Results demonstrated that low FAM99B expression was associated with vascular invasion (P = 0.001), advanced histologic grade (P = 0.001), and T stage (P = 0.045). However, no significant difference was observed in other clinicopathological features such as age, gender, race, tumor status, hepatitis B virus infection, pathologic stage, N stage, M stage, and Child–Pugh classification grade (P > 0.05; Table 1). Kaplan–Meier analysis suggested that lower FAM99B expression was associated with a poor OS for patients with HCC (χ2 = 4.295, P = 0.038; Fig. 1c). However, no significant association was found between FAM99B expression and disease-free survival (DFS) (χ2 = 0.030, P = 0.863; Fig. 1d).

Expression level and survival analysis of FAM99B in patients with HCC based on TCGA cohort. a FAM99B expression in HCC tissues and adjacent normal tissues. b ROC curve analysis of the diagnostic efficiency of FAM99B expression in HCC. c Correlations between FAM99B expression and the overall survival (OS) of HCC patients. d Correlations between FAM99B expression and disease-free survival (DFS) of HCC patients
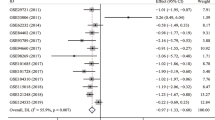
Meta-analysis of FAM99B expression in HCC based on GEO data sets
Nine GEO data sets (GSE27462, GSE29721, GSE33006, GSE49713, GSE50579, GSE57555, GSE62232, GSE65485, and GSE84402) met the inclusion criteria and were enrolled in our study. The normalized FAM99B expression level between HCC and noncancerous tissues in GEO data sets is visualized in Fig. 2a–i. The basic information and normalized FAM99B expression level are shown in Table 2. To mediate the small sample effect of individual data sets, we pooled all six data sets to draw a credible conclusion. A random-effect model was applied, and the combined result indicated that FAM99B was downregulated in HCC tissues (SMD = − 1.07, 95% CI (− 1.66, − 0.49); P < 0.001; Fig. 2j). There was significant heterogeneity among these studies (I2 = 59.8%; P = 0.011). No significant publication bias was observed (Begg’s test, P = 0.602; Fig. 2k) among the data sets. Moreover, the result of sensitivity analyses (Fig. S1) indicated that our meta-analysis was stable.

Relative expression level of FAM99B between HCC tissues and adjacent normal tissues in GEO database. a–i FAM99B expression in nine GEO data sets. j Forest plot of nine GEO data sets for the FAM99B expression in HCC (with random-effect model). k Funnel plot of Begg’s test to assess the publication bias of these nine GEO data sets
FAM99B displays a low expression level in HCC tissues based on HCC cohort from Guangxi
To validate the relationship between FAM99B and HCC, we further detected the expression level of FAM99B in 80 pairs of HCC tissues and their corresponding adjacent normal tissues using qRT-PCR for the HCC cohort from Guangxi Medical University. The results indicated that FAM99B displayed a lower expression level in 96.25% (77/80) of HCC tissues compared with their corresponding adjacent normal tissues (P < 0.001; Table 3; Fig. 3a, b). Moreover, the ROC curve analysis revealed that FAM99B had significant performance in discriminating HCC tissues from normal tissues (AUC = 0.707, 95% CI 0.630–0.776, P < 0.0001). With a cutoff value of 0.6404, the sensitivity and specificity were 70.0% and 63.7%, respectively (Fig. 3c). To explore the clinical significance of FAM99B in HCC, patients were also divided into a high expression group and low expression group according to the median value (0.3593) of FAM99B expression. Consistent with TCGA results, patients with low FAM99B expression were associated with microvascular invasion (P = 0.014). However, no significant correlation was found in age, gender, liver cirrhosis, hepatitis B virus infection, tumor number, tumor size, tumor invasion, lymph node metastasis, Child–Pugh classification grade, BCLC stage, and Edmondson stage group (P > 0.05; Table 3).

Expression level of FAM99B in Guangxi HCC cohort. a FAM99B was downregulated in HCC tissues. b Paired t test of FAM99B expression in HCC tissues in comparison with corresponding adjacent normal tissues. c ROC curve analysis of the diagnostic efficiency of FAM99B expression in HCC
Overexpression of FAM99B inhibits cell proliferation, migration, and invasion of HCC cells
To understand the biological function of FAM99B in HCC cell lines, we first examined the expression of FAM99B using qRT-PCR. We found that cells (HCCLM3, MHCC97L, MHCC97H) with metastatic potential have a lower FAM99B expression level than those (Huh-7, HepG2, Hep3B) with little or no metastatic potential (Fig. 4a). HCCLM3 was then selected to construct a stable FAM99B overexpression cell line. Transfection results showed that the level of FAM99B expression was remarkably increased in the Lv-FAM99B group compared to the Lv-NC group in HCCLM3 cells (Fig. 4b). CCK-8 assay showed that increased expression of FAM99B could remarkably inhibit cell proliferation (Fig. 4c). Further evidence can be observed in the results of the colony formation assay, which demonstrated that FAM99B overexpression decreased colony counts in HCC cells (Fig. 4d). The inverse correlation between FAM99B expression and vascular invasion prompted us to investigate whether FAM99B affects cell migration and invasion ability. As expected, transwell migration and invasion assays showed that enhanced FAM99B expression significantly decreased the cell migration and invasion capabilities of HCC cells compared to the Lv-NC group (Fig. 4e). The above results suggest that elevated FAM99B expression levels repress cell proliferation and invasion in HCC cells.

FAM99B suppress cell proliferation, migration, and invasion of HCC cells. a Expression level of FAM99B in HCC cell lines. b FAM99B was significantly up-regulated in Lv-FAM99B group in HCCLM3. c CCK-8 assay exhibited inhibition of cell proliferation caused by overexpression of FAM99B. d Colony formation assay showed that reduced colonies was observed in Lv-FAM99B group. e Transwell assays indicated that the migration and invasion abilities were suppressed by enhanced FAM99B. The above assays were performed in triplicates (*P < 0.05; **P < 0.01; ***P < 0.001)
Co-expressed genes of FAM99B and pathway analyses
To further investigate the underlying molecular mechanism of FAM99B in HCC, the co-expressed genes of FAM99B were predicted using three lncRNA-related prediction websites. A total of 4934 co-expressed genes were identified in the cBioPortal database, and more than 5000 genes were found in the MEM database with statistical significance; by contrast, only 464 related genes were gathered in the TANRIC database. The top 1000 most significant co-expressed genes were extracted from the cBioPortal and MEM database for further analysis. Finally, 174 intersected genes were identified in at least two databases (Fig. 5a). Subsequently, these 174 genes were chosen to perform GO annotation and pathway analyses using KOBAS database. For GO annotation analysis, the top 20 GO terms are listed in Table 4. The results of KEGG pathway analysis showed that the potentially related genes were mainly enriched in the pathways “Metabolic pathways”, “Fatty acid elongation”, and “Complement and coagulation cascades”. As for the PANTHER analysis, co-expressed genes were significantly involved in the “Blood coagulation” and “Nicotine degradation” pathways (Table 5; Fig. 5b).

Bioinformatics analysis to investigate the underlying molecular mechanism of FAM99B in HCC. a Venn diagram of the overlapping genes among the predicted co-expressed genes of FAM99B using the cBioPortal (http://www.cbioportal.org/), MEM (https://biit.cs.ut.ee/mem/), and TANRIC (https://ibl.mdanderson.org/tanric/_design/basic/index.html) databases. b Significant enrichment pathways in the KEGG and PANTHER pathway analysis using KOBAS 3.0 (http://kobas.cbi.pku.edu.cn/)
Discussion
The present study, to the best of our knowledge, is the first to investigate the association between FAM99B and HCC. After screening the expression levels of FAM99B in TCGA and GEO databases, we found that FAM99B was significantly downregulated in HCC tissues compared to adjacent normal tissues. This was further verified through result obtained from our cohort. An ROC curve analysis indicated that FAM99B had a high diagnostic efficiency in differentiating HCC tissues. Results indicated that low expression of FAM99B was associated with vascular invasion, advanced histologic grade, and T stage. Survival analysis of TCGA data revealed that decreased expression of FAM99B was significantly associated with poor OS. On the contrary, overexpression of FAM99B inhibited cell proliferation, migration, and invasion of HCC cells in vitro. Bioinformatics analysis showed that FAM99B may participate in metabolic and blood coagulation pathways in HCC. Collectively, the above results identified a tumor-suppressing role of FAM99B in HCC.
Patients with HCC are often only diagnosed at an advanced stage, and this stage is usually accompanied with tumor metastasis (Zhou 2002). Currently, AFP is the most clinically recognized diagnostic biomarker of HCC with a high specificity of 95% (87–98%), but its sensitivity of 66% (53–76%) is rather low (Wan et al. 2014). Therefore, finding a suitable auxiliary diagnostic biomarker in HCC is crucial. In the present study, we found that FAM99B had high performance in diagnosing HCC tissues with a sensitivity of 71.97% and specificity of 92.00% in TCGA data. Moreover, our qRT-PCR result in 80 sample pairs showed a sensitivity of 70.0% and a specificity of 63.7%. According to RNA-seq data in 53 human normal tissues, we recognized that FAM99B is a liver-enriched lncRNA (Fagerberg et al. 2014). In addition, decreased levels of FAM99B were significantly correlated with malignant phenotypes and poor OS in patients with HCC. Thus, FAM99B may serve as a promising specific auxiliary diagnostic biomarker for tumor progression and prognosis prediction of HCC patients.
In this study, we also found that overexpression of FAM99B resulted in phenotype changes in HCC cell lines, including repressed cell proliferation, inhibited cell migration, and invasion ability. Reports have proven that cancer cells with high migration and invasion activities often lead to the poor survival of patients, which correlates with the aggressiveness and recurrence of HCC (Ge et al. 2017; Liu et al. 2017; Yuan et al. 2012). Compared to protein-coding genes, lncRNAs are more tissue-specific, and thus are expected to be more promising prognostic indicators and therapeutic targets (Gutschner and Diederichs 2012). PCA3, a prostate-specific lncRNA which was significantly up-regulated in prostate cancer, has proved to be a better diagnostic biomarker than prostate-specific antigens due to its higher diagnostic accuracy (Lee et al. 2011). As for liver-enriched lncRNAs, LINC01093 could suppress HCC progression through inhibition of cell proliferation and metastasis (He et al. 2019). Another lncRNA, lnc18q22.2, has been shown to play a role in cell viability in human non-alcoholic steatohepatitis (Atanasovska et al. 2017). Therefore, it is particularly important to discover more liver-specific lncRNAs associated with HCC progression to disclose the molecular mechanisms of HCC-specific lncRNAs.
The results of KEGG pathway analysis exhibited that related genes of FAM99B mainly participated in “Metabolic pathways” and “Fatty acid elongation” pathways. These findings seem to be biologically plausible. Recent studies have demonstrated that alterations of metabolic pathways can provide energy and nutrients for the growth and proliferation of tumor cells (DeBerardinis et al. 2008; Vander Heiden et al. 2011), and contribute to the epithelial-to-mesenchymal transition (EMT) process in cancer (Sciacovelli and Frezza 2017). Kessler SM et al. (Kessler et al. 2014) reported that fatty acid elongation contributes to the progression of non-alcoholic steatohepatitis-related HCC. For PANTHER pathway analysis, blood coagulation was the most significant enrichment pathway which has been proven to be closely associated with tumor development by supporting the invasive growth of neoplastic tissues and blood vessels (Boccaccio and Medico 2006). Hence, our findings that confirm FAM99B could repress cell proliferation, migration, and invasion in vitro is consistent with the results of the pathway enrichment. The pathway analyses could be regarded as a preliminary study to explore the underlying molecular mechanism of FAM99B in HCC. To illuminate the specific mechanism of its association with HCC, further in vitro and vivo studies are warranted.
In summary, our comprehensive research indicates that FAM99B is downregulated in HCC tissues and is negatively correlated with HCC progression and prognosis. Overexpression of FAM99B could inhibit cell proliferation, migration, and invasion in HCC cell lines. Bioinformatics analysis suggests that FAM99B may serve as a tumor suppressor by regulating some important pathways in HCC. These results will improve our understanding on the molecular mechanisms of HCC and may contribute to early diagnosis and prognosis prediction.
References
Adler P, Kolde R, Kull M, Tkachenko A, Peterson H, Reimand J, Vilo J (2009) Mining for coexpression across hundreds of datasets using novel rank aggregation and visualization methods. Genome Biol 10:R139. https://doi.org/10.1186/gb-2009-10-12-r139
Atanasovska B et al (2017) A liver-specific long noncoding RNA with a role in cell viability is elevated in human nonalcoholic steatohepatitis. Hepatology 66:794–808. https://doi.org/10.1002/hep.29034
Begg CB, Mazumdar M (1994) Operating characteristics of a rank correlation test for publication bias. Biometrics 50:1088–1101
Boccaccio C, Medico E (2006) Cancer and blood coagulation. Cell Mol Life Sci 63:1024–1027. https://doi.org/10.1007/s00018-005-5570-9
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394–424. https://doi.org/10.3322/caac.21492
Cerami E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404. https://doi.org/10.1158/2159-8290.CD-12-0095
Chang J et al (2004) miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol 1:106–113
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7:11–20. https://doi.org/10.1016/j.cmet.2007.10.002
DerSimonian R, Laird N (2015) Meta-analysis in clinical trials revisited. Contemp Clin Trials 45:139–145. https://doi.org/10.1016/j.cct.2015.09.002
Fagerberg L et al (2014) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteom 13:397–406. https://doi.org/10.1074/mcp.M113.035600
Forner A, Reig M, Bruix J (2018) Hepatocellular carcinoma. Lancet 391:1301–1314. https://doi.org/10.1016/S0140-6736(18)30010-2
Gao J et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:l1. https://doi.org/10.1126/scisignal.2004088
Ge Z et al (2017) Long noncoding RNA SchLAH suppresses metastasis of hepatocellular carcinoma through interacting with fused in sarcoma. Cancer Sci 108:653–662. https://doi.org/10.1111/cas.13200
Gutschner T, Diederichs S (2012) The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol 9:703–719. https://doi.org/10.4161/rna.20481
He Y, Meng XM, Huang C, Wu BM, Zhang L, Lv XW, Li J (2014) Long noncoding RNAs: novel insights into hepatocelluar carcinoma. Cancer Lett 344:20–27. https://doi.org/10.1016/j.canlet.2013.10.021
He J et al (2019) A novel, liver-specific long noncoding RNA LINC01093 suppresses HCC progression by interaction with IGF2BP1 to facilitate decay of GLI1 mRNA. Cancer Lett 450:98–109. https://doi.org/10.1016/j.canlet.2019.02.033
Huang S, He X (2011) The role of microRNAs in liver cancer progression. Br J Cancer 104:235–240. https://doi.org/10.1038/sj.bjc.6606010
Kessler SM et al (2014) Fatty acid elongation in non-alcoholic steatohepatitis and hepatocellular carcinoma. Int J Mol Sci 15:5762–5773. https://doi.org/10.3390/ijms15045762
Lanzafame M, Bianco G, Terracciano LM, Ng CKY, Piscuoglio S (2018) The role of long non-coding RNAs in hepatocarcinogenesis. Int J Mol Sci. https://doi.org/10.3390/ijms19030682
Lee GL, Dobi A, Srivastava S (2011) Prostate cancer: diagnostic performance of the PCA3 urine test. Nat Rev Urol 8:123–124. https://doi.org/10.1038/nrurol.2011.10
Li J et al (2015) TANRIC: an interactive open platform to explore the function of lncRNAs in cancer. Cancer Res 75:3728–3737. https://doi.org/10.1158/0008-5472.can-15-0273
Liu J, Lu C, Xiao M, Jiang F, Qu L, Ni R (2017) Long non-coding RNA SNHG20 predicts a poor prognosis for HCC and promotes cell invasion by regulating the epithelial-to-mesenchymal transition. Biomed Pharmacother 89:857–863. https://doi.org/10.1016/j.biopha.2017.01.011
Ni W et al (2017) A novel lncRNA uc.134 represses hepatocellular carcinoma progression by inhibiting CUL4A-mediated ubiquitination of LATS1. J Hematol Oncol 10:91. https://doi.org/10.1186/s13045-017-0449-4
Sciacovelli M, Frezza C (2017) Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J 284:3132–3144. https://doi.org/10.1111/febs.14090
Sun QM et al (2018) Long non-coding RNA00607 as a tumor suppressor by modulating NF-kappaB p65/p53 signaling axis in hepatocellular carcinoma. Carcinogenesis. https://doi.org/10.1093/carcin/bgy113
Vander Heiden MG et al (2011) Metabolic pathway alterations that support cell proliferation. Cold Spring Harb Symp Quant Biol 76:325–334. https://doi.org/10.1101/sqb.2012.76.010900
Wan HG, Xu H, Gu YM, Wang H, Xu W, Zu MH (2014) Comparison osteopontin vs AFP for the diagnosis of HCC: a meta-analysis. Clin Res Hepatol Gastroenterol 38:706–714. https://doi.org/10.1016/j.clinre.2014.06.008
Wang HP, Rogler CE (1988) Deletions in human chromosome arms 11p and 13q in primary hepatocellular carcinomas. Cytogenet Cell Genet 48:72–78. https://doi.org/10.1159/000132593
Wang H et al (2018) Long noncoding RNA miR503HG, a prognostic indicator, inhibits tumor metastasis by regulating the HNRNPA2B1/NF-kappaB pathway in hepatocellular carcinoma. Theranostics 8:2814–2829. https://doi.org/10.7150/thno.23012
Xie C et al (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39:W316–W322. https://doi.org/10.1093/nar/gkr483
Yao J et al (2017) Profiling, clinicopathological correlation and functional validation of specific long non-coding RNAs for hepatocellular carcinoma. Mol Cancer 16:164. https://doi.org/10.1186/s12943-017-0733-5
Yuan SX et al (2012) Long noncoding RNA associated with microvascular invasion in hepatocellular carcinoma promotes angiogenesis and serves as a predictor for hepatocellular carcinoma patients’ poor recurrence-free survival after hepatectomy. Hepatology 56:2231–2241. https://doi.org/10.1002/hep.25895
Zhou XD (2002) Recurrence and metastasis of hepatocellular carcinoma: progress and prospects. Hepatobil Pancreat Dis Int 1:35–41
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81660563 and 81402756).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that there are no conflicts of interest in this study.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mo, M., Liu, S., Ma, X. et al. A liver-specific lncRNA, FAM99B, suppresses hepatocellular carcinoma progression through inhibition of cell proliferation, migration, and invasion. J Cancer Res Clin Oncol 145, 2027–2038 (2019). https://doi.org/10.1007/s00432-019-02954-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-019-02954-8