
Abstract
Purpose
To evaluate a new strategy for profiling proteomic changes in colorectal cancer (CRC).
Methodes
We used laser capture microdissection (LCM) to obtain cells from 20 CRC and paired normal mucosal tissues. The differential proteins between the microdissected tumor cells and normal mucosa epithelia were analyzed by acetylation stable isotopic labeling coupled with L linear ion trap Fourier transform ion cyclotron resonance mass spectrometry (LTQ-FT MS). Western blotting was used to assess the differential expression of proteins. We used bioinformatics tools for cluster and ingenuity pathway analysis of the differential proteins.
Results
In total, 798 confident proteins were quantified and 137 proteins were differentially expressed by at least twofold, including 67 that were upregulated and 70 that were downregulated in cancer. Two differential proteins, solute carrier family 12 member 2 (SLC12A2) and Ras-related protein Rab-10, were validated by Western blotting, and the results were consistent with acetylation stable isotopic labeling analysis. According to gene ontology analysis, CRC-related differential proteins covered a wide range of subcellular locations and were involved in many biological processes. According to ingenuity pathway analysis of the differential proteins, the most relevant canonical pathway associated with CRC was the 14-3-3-mediated signaling pathway, and seven reliable functional networks including cellular growth and proliferation, amino acid metabolism, inflammatory response, embryonic development, carbohydrate metabolism, cellular assembly and organization, and cell morphology were obtained.
Conclusions
Combination of LCM, acetylation stable isotopic labeling analysis and LTQ-FT MS is effective for profiling proteomic changes in CRC cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is one of the most commonly diagnosed cancers and is listed as the third leading cause of cancer-related deaths worldwide (Siegel et al. 2014). Treatments for CRC have made great progresses in recent years (Cunningham et al. 2004; Berretta et al. 2008; Douillard et al. 2000). However, the responses to these treatments are not always satisfactory. Thus, improved knowledge of cancer biology is required to understand the diversity of CRC patients so as to guide treatment and improve the outcome of patients (Popa-Velea et al. 2010). The development of proteomics technology will help to provides deeper insights into the biology of CRC (Hanash 2003).
Surgical tissue specimens are ideal for searching for protein markers through proteomic analysis. However, solid tissues are complex structures composed of heterogeneous mixtures of morphologically and functionally distinct cell types. Analysis of whole tumor tissue samples complicates the identification of tumor markers and may arouse controversy (Li et al. 2007). Therefore, it is essential to analyze specific cell types such as tumor cells or normal epithelial cells to identify and define biologically important processes. By isolating pure tumor cells, laser capture microdissection (LCM) allows cell type-specific molecular analysis of tumor tissues and provides new insights into normal cell biology and pathogenic mechanisms (Datta et al. 2015).
LCM has been widely used in comparative genomics and is an attractive prospect in proteomics (Yonemori et al. 2013). Unfortunately, the number of targeted cells procured by LCM is generally small and the process of harvesting enough cells for analysis is laborious and time-consuming, which limits the differential proteome analysis by conventional gel-based proteomics platforms such as 2-D gel electrophoresis (2-DE) and 2-D differential gel electrophoresis (2D-DIGE; Cheng et al. 2008). Recently, a quantitative proteomics strategy based on stable isotopic labeling combined with linear ion trap Fourier transform ion cyclotron resonance mass spectrometry (LTQ-FT MS) has been approved as a valuable and reliable differential proteomic analysis strategy. This strategy can quantitatively analyze samples with high precision and screen differentially expressed proteins in high throughput simultaneously (Umar et al. 2007).
In this study, LCM was used to harvest cells from 20 CRC and paired normal mucosal tissues. Protein expression patterns were compared between tumor cells and normal epithelial cells by means of d0/d3 acetylation labeling coupled with LTQ-FT MS. Our study indicates that our proteomic strategy possesses obvious advantages of allowing analysis of small samples and providing high proteome coverage for complex biological samples, and provides a useful resource for CRC proteomics.
Materials and methods
Clinical samples
This study included tumor and paired normal tissues from 20 randomly selected patients with CRC who underwent curative surgery at the Peking University People’s Hospital (PUPH), China. The clinical and pathological data for the patients are summarized in Online Resource 1 Table S1. None of the patients received neoadjuvant chemotherapy before surgery. Tumor and normal tissues were obtained at the time of surgery. Each sample was divided into two parts: One was immediately snap frozen in liquid nitrogen and then stored at −80 °C for LCM or Western blotting; the other was fixed in 10 % formaldehyde solution for hematoxylin and eosin (HE) staining. This study was approved by the Ethics Committee of PUPH, and informed consent was obtained from all participants.
LCM and protein extraction
LCM was performed with a laser microdissection and pressure catapulting microscope (Palm, Bernried, Germany). One 4-µm and four 8-µm thick frozen sections were prepared from fresh tissues. The 4-µm sections were stained with HE for microscopic observation and location of the desired cells, and the 8-µm thick sections were subjected to LCM (Fig. 1), approximately 3000 cells in each section for a maximum of 30 min. Equal microdissected cells of each sample from tumor tissues or matched normal mucosa were pooled. The proteins were extracted from the pooled cells using a urea lysis buffer containing 7 M urea, 2 M thiourea, 100 mM DTT, 4 % CHAPS, and 1× protease inhibitor cocktail (Roche, Penzberg, Germany) and stored at −80 °C until use. Protein concentrations were measured in duplicate with the Bradford method.

LCM of normal mucosa (a, c, e) and CRC (b, d, f). a, d 4 µm frozen sections stained with HE; b, e 10 µm unstained frozen sections before LCM; c, f 10 µm unstained frozen sections after LCM. Magnification ×100
Protein separation and in-gel digestion
Equal amounts of protein (20 µg) from noncancerous epithelial cells and adenocarcinoma cells were boiled in SDS-PAGE sample buffer, separated on the same 12 % SDS-PAGE and stained with Coomassie Brilliant Blue. Each gel lane was cut into 10 sections for in-gel microwave-assisted tryptic digestion according to Sun et al. (2006). The excised gel strips were chopped into ~1-mm3 pieces and destained twice with 50 % acetonitrile (ACN)/25 mM ammonium bicarbonate solution. After dehydration in 100 % ACN for 10 min, the supernatants were discarded. The gel pieces were rehydrated with trypsin solution (0.01 µg/µL trypsin in 25 mM ammonium bicarbonate) on ice for 20 min and then digested in a microwave oven at 750 W for 8 min, followed by incubation overnight at 37 °C. The supernatants were collected, and the tryptic peptides were extracted from the gel sequentially with 5 % trifuloroacetic acid (TFA) in the microwave oven at 750 W for 8 min, and with 2.5 % TFA, 50 % ACN in the microwave oven at 750 W for 8 min. The extracts were dried completely by centrifugal lyophilization for further stable isotopic labeling.
Acetylation stable isotopic labeling
Gel pieces from normal epithelial cells were labeled with d0 acetylation of the N termini, while the CRC samples were labeled with d3 acetylation. The labeling was performed according to Everley et al. (2006) and Nuan et al. (2007). One hundred microliters of 1 M O-methylisourea (dissolved in 0.5 M carbonate buffer, adjusted to pH 11 with 1 M NaOH) was added to the resultant peptides from gel digestion and incubated at 37 °C for 2 h. The solutions were adjusted back to pH 8.0 with 1 M HCl for acetylation. Another 200 µg borate buffer was added to the solution for the maintenance of pH value. D0-acetic anhydride and d3-acetic anhydride were diluted in 1 M tetrahydrofuran. The amount of diluted acetic anhydride was 1 µL per 50 µg digested peptides. The acetylation reaction was preceded at room temperature for 1 h. N-Hydroxylamine was used to hydrolyze the esters formed during the acetylation reaction incubating for 30 min (pH 11.0). Heavy-labeled (d3) samples were mixed with the corresponding light-labeled (d0) samples, respectively, desalted using the solid phase extraction (Sigma, St Louis, MO, USA) column lyophilized, and stored at −80 °C before use.
Protein identification and quantification
Ten labeled sample pairs were analyzed using LTQ-FT MS (Thermo Electron, San Jose, CA, USA) with an Agilent 1100 nano-flow liquid chromatography system. The LTQ-FT MS data were searched against the IPI human 3.23 using local MASCOT (version 1.9) with a 95 % confidence level. Based on the MASCOT results, the ratios of the monoisotopic peaks between the light-labeled peptides and heavy-labeled peptides were automatically calculated using an in-house program and the protein ratios were calculated from the average of all quantified peptides. The detailed information is shown in Online Resource 2 Materials and methods S1.
Western blotting
The expression levels of two dysregulated proteins in the microdissected samples for acetylation labeling were validated in the whole lysates of paired CRC and adjacent normal mucosa samples to confirm the results of the proteomic approach. Primary antibodies were purchased from the suppliers: Ras-related protein Rab-10 (RAB10; rabbit polyclonal; 1:1000 dilution) from Proteintech (Chicago, IL, USA) and solute carrier family 12 member 2 (SLC12A2; chicken polyclonal; 1:1000 dilution) from GenWay Biotech (San Diego, CA, USA). To perform Western blotting, 25 μg protein was separated by 12 % SDS-PAGE. Then proteins were transferred to nitrocellulose membranes (Amersham Biosciences, Little Chalfont, UK). The membranes were blocked with 5 % nonfat dried milk in PBS containing 0.05 % Tween-20 for 1 h at room temperature, and incubated with the primary antibodies for 2 h at room temperature, or overnight at 47 °C. After washing three times for 5 min each, the membranes were incubated with the secondary antibody (1:10 000 dilution; Jackson, West Grove, PA, USA) for 1 h at room temperature. Finally, target proteins were detected by ECL (Pierce, Rockford, USA) and quantitated by densitometry using ImageQuant image analysis system (Storm Optical Scanner; Molecular Dynamics, Eugene, OR, USA). β-Actin (mouse monoclonal; 1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used simultaneously as a loading control.
Analysis of differential proteins
The differential proteins in CRC and normal epithelial cells were analyzed by informatics tools. Gene Ontology (GO, www.geneontology.org) was used to classify the differential proteins (ratios were ≥2.0 or ≤0.5) according to different subcellular location and biological function. Ingenuity Pathways Analysis (IPA, Ingenuity Systems, Redwood City, CA, USA, www.ingenuity.com) was applied for identifying the statistically relevant (P < 0.05) canonical pathways and functional networks related to the differential proteins.
Statistical analysis
All statistical analyses were performed using SPSS 19.0 software (SPSS, Inc., Chicago, IL, USA). A t test was used to assess the differences of protein expression between tumor and normal mucosa.
Results
Protein identification and quantification
In total, 798 confident proteins were identified and quantified. One hundred and thirty-seven proteins showed at least a twofold difference in expression level in CRC. Sixty-seven proteins were upregulated (ratios ≥2.0), and 70 were downregulated (ratios ≤0.5) in CRC cells compared with normal epithelial cells (Online Resource Table S2).
Western blotting
To validate the results measured by d0/d3 acetylation analysis, we performed Western blotting with two protein antibodies in the 20 paired CRC samples.
Expression of RAB10 and SLC12A2 was higher in pooled tumor cells than pooled normal epithelial cells by 3.2-fold and 3.0-fold, respectively, according to the acetylation stable isotopic labeling method coupled with LTQ-FT MS. Through validation in the 20 paired CRC samples, significant differences were observed between tumor and normal mucosa concerning both RAB10 and SLC12A2. Sixteen cases showed an increase in RAB10 (Fig. 2), and 15 showed an increase in SLC12A2 (Fig. 3) in cancer tissues.

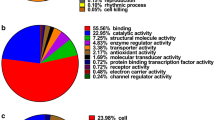
Cellular categorization of CRC-related differential proteins based on the annotations of GO. a Subcellular localization of differential proteins. b Function distribution of differential proteins

Expression of RAB-10 in 20 paired samples of CRC (T) and normal mucosa (N). The intensity of each band was measured with ImageQuant image analysis system and normalized with β-actin. a Expression of RAB-10 was significantly upregulated in tumor tissues compared with normal mucosa. b The difference in RAB-10 expression between (T) and (N) was evaluated by t test
Analysis of differentially expressed proteins
According to GO analysis, the 137 differentially expressed proteins (ratios ≥2.0 or ≤0.5) covered almost all categories of subcellular localization and a variety of biological processes. Most differential proteins were located in the cytoplasm, plasma membrane, nucleus and mitochondrial membrane (Fig. 4a). With regard to functional distribution, the two major groups of proteins were related to signal transduction and molecular transport (Fig. 4b).

Expression of SLC12A2 in 20 paired samples of CRC (T) and normal mucosa (N). The intensity of each band was measured with ImageQuant image analysis system and normalized with β-actin. a Expression of SLC12A2 was significantly upregulated in tumor tissues compared with normal mucosa. b The difference of SLC12A2 expression between (T) and (N) was evaluated by t test
IPA of the differential proteins showed that multiple canonical signaling pathways were significantly changed in CRC (Table 1). The 14-3-3-mediated signaling pathway was the most significant. According to network analysis, seven reliable functional networks including cell growth and proliferation (containing 25 differential proteins, Fig. 5a), amino acid metabolism (containing 16 differential proteins, Fig. 5b), inflammatory response (containing 15 differential proteins, Fig. 5c), embryonic development (containing 15 differential proteins, Fig. 5d), carbohydrate metabolism (containing 14 differential proteins, Fig. 5e), cellular assembly and organization (containing 13 differential proteins, Fig. 5f), and cell morphology (containing 10 differential proteins, Fig. 5g) were obtained.

Biological network generated by IPA for differentially expressed proteins between CRC and normal mucosa. Red and green represent up- and down-regulated proteins identified in this work; white indicates proteins that were not in the differentially expressed protein lists but related to the network. Solid arrows represent known direct interactions, and dotted arrows mean indirect interactions
Discussion
Several tumor markers such as carcinoembryonic antigen, carbohydrate antigen (CA)19-9), CA24-2, CA50, CA72-4 and tissue polypeptide antigen have been commonly used in the diagnosis of CRC. However, clinical application of these markers is limited by their lack of specificity and sensitivity (Grotowski 2002; Fakih and Padmanabhan 2006; Nicolini et al. 2010; Carpelan-Holmström et al. 2004). With the rise of proteomics, oncologists have been actively researching CRC-associated protein markers.
Proteomics analysis of CRC has yielded some meaningful results, but has yet to find any biomarkers specific and sensitive enough to be used in clinical practice, which may be attributed to the fact that traditional proteomics platforms (e.g., 2-DE and 2D-DIGE) used in previous studies had obvious limitations such as: poor repeatability and severe bias; only a limited number of the differentially expressed proteins could be detected; and yet some important molecular markers were missed (Gygi et al. 2000). Therefore, it is necessary to improve the methods in studies of tumor markers.
In this study, we applied LCM and d0/d3 acetylation isotopic labeling combined with LTQ-FT MS to the analysis of differentially expressed proteins in CRC for the first time. With only 20 μg proteins obtained by LCM, we identified and quantified 798 confident proteins, including 137 that were significantly differentially expressed (ratio ≥2 or ≤0.5) between colorectal adenocarcinoma and normal mucosa. Among the differentially expressed proteins, many dysregulated proteins in CRC, such as heat shock protein 10, S100A6, mortalin, annexin A4, alpha 1 antitrypsin, and selenium binding protein 1, have shown consistency with preceding proteomic research on CRC (Melle et al. 2006; Dundas et al. 2005; Duncan et al. 2008; Stulík et al. 2000; Li et al. 2008; Bi et al. 2006). Apart from the differential proteins reported by other authors, we also found many new proteins with no relevant research reports, or of unknown function in CRC. These proteins may be related to the occurrence and development of CRC, which could provide abundant resources for screening of CRC tumor markers and investigating the underlying mechanism of carcinogenesis. Compared with 650 μg protein samples needed for the strategy combining LCM and conventional gel-based proteomics platforms (Cheng et al. 2008), only a small amount of protein samples (20 μg) was enough for our strategy owing to high precision and high sensitivity of LTQ-FT MS. However, LCM is rather complicated and time-consuming (30 min or more), it is maybe impossible that this method will be generally and widely used for many samples especially in protein analysis.
GO cellular localization analysis showed that the differentially expressed proteins were distributed in almost all categories of subcellular localizations including cytoplasm (16 %), cell membrane (12 %), mitochondria (12 %), nuclei (7 %), and cytoskeleton (7 %), which shows the advantage of our strategy in tumor biomarker searching with better proteome coverage to some extent. The results also implied that it is necessary to carry out research on tumor markers of CRC and related carcinogenic mechanisms at the subcellular level. GO function analysis showed that the differentially expressed proteins were involved in a variety of cellular biological processes, including signal transduction, molecular transport, differentiation, proliferation, cell cycle, and material metabolism. Among which, signal transduction-related proteins (15 %) was the most important functional category. Disorders in the signal transduction system may cause abnormal expression of genes that play a key role in the occurrence and development of cancer (Krausova and Korinek 2012). Rab proteins, one of the Ras superfamily members of monomeric GTPases, play important role in regulating growth factor signaling to PI3 K/AKT (Wheeler et al. 2015). In this study, we found RAB-10, one member of Rab family, was upregulated significantly in CRC for the first time, which indicates RAB-10 may have latent oncogenic potential in CRC carcinogenesis through PI3 K/AKT signaling. Molecular transport proteins (12 %) were the second most important functional category of differential proteins we found. As a sodium–potassium-chloride cotransporter,SLC12A2 plays an important role in the regulation of cell volume and contributes to cancerous features such as proliferation, migration and invasion (Becchetti et al. 2013; Haas and Sontheimer 2010). Upregulated expression of SLC12A2 indicates that important changes in ion transport occur during the process of CRC and may serve as a potential anticancer target.
In this study, IPA of the differential proteins between CRC and normal mucosa showed that the most relevant canonical pathway associated with CRC was the 14-3-3-mediated signaling pathway including molecules such as tubulin beta-4, 14-3-3 theta, 14-3-3 gamma, 14-3-3 beta, 14-3-3 eta and tubulin beta-1. 14-3-3 proteins are a family of conserved regulatory molecules expressed in all eukaryotic cells. 14-3-3 proteins have the ability to regulate the ERK/MAPK pathway and the PI3 K/AKT pathways, which are key growth-promoting pathways associated with the ability of cancer cells to proliferate in an uncontrolled manner (Panagopoulos et al. 2013). It has been demonstrated that expression of 14-3-3 proteins is significantly elevated in several types of cancer (Wang et al. 2011; Murata et al. 2012). Our results suggest that the 14-3-3-mediated signaling pathway has important roles in CRC and calls for further study. Seven functional networks with high confidence are represented by network analysis, which are related to cellular growth and proliferation, amino acid metabolism, mechanism of infection, embryonic development, carbohydrate metabolism, cellular assembly and organization, and cell morphology. These results showed that the carcinogenesis and development of CRC is a complicated process, involving changes in a variety of signaling pathways and complex functional networks. Further analysis of the key molecules in different pathway networks may provide important information for the interpretation of the pathogenesis of CRC.
In conclusion, this study revealed many novel differential proteins in CRC compared with normal mucosa, in addition to many previously reported CRC-associated proteins. Abnormality of the 14-3-3-mediated signaling pathway was an important event in the carcinogenesis and development of CRC. We showed that the strategy combining LCM, acetylation stable isotopic labeling analysis and LTQ-FT MS were effective for searching for novel biomarkers and profiling proteomic alterations in CRC.
References
Becchetti A, Munaron L, Arcangeli A (2013) The role of ion channels and transporters in cell proliferation and cancer. Front Physiol 4:312
Berretta M, Lleshi A, Zanet E et al (2008) Bevacizumab plus irinotecan-, fluorouracil-, and leucovorin-based chemotherapy with concomitant HAART in an HIV-positive patient with metastatic colorectal cancer. Onkologie 31:394–397
Bi X, Lin Q, Foo TW et al (2006) Proteomic analysis of colorectal cancer reveals alterations in metabolic pathways: mechanism of tumorigenesis. Mol Cell Proteom 5:1119–1130
Carpelan-Holmström M, Louhimo J, Stenman UH et al (2004) CEA, CA 242, CA 19-9, CA 72-4 and hCG beta in the diagnosis of recurrent colorectal cancer. Tumour Biol 25:228–234
Cheng AL, Huang WG, Chen ZC et al (2008) Identificating cathepsin D as a biomarker for differentiation and prognosis of nasopharyngeal carcinoma by laser capture microdissection and proteomic analysis. J Proteome Res 7:2415–2426
Cunningham D, Humblet Y, Siena S et al (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 351:337–345
Datta S, Malhotra L, Dickerson R et al (2015) Laser capture microdissection: big data from small samples. Histol Histopathol 30:1255–1269
Douillard JY, Cunningham D, Roth AD et al (2000) Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 355:1041–1047
Duncan R, Carpenter B, Main LC et al (2008) Characterization and protein expression profiling of annexins in colorectal cancer. Br J Cancer 98:426–433
Dundas SR, Lawrie LC, Rooney PH, Murray GI (2005) Mortalin is over-expressed by colorectal adenocarcinomas and correlates with poor survival. J Pathol 205:74–81
Everley PA, Bakalarski CE, Elias JE et al (2006) Enhanced analysis of metastatic prostate cancer using stable isotopics and high mass accuracy instrumentation. J Proteome Res 5:1224–1231
Fakih MG, Padmanabhan A (2006) CEA monitoring in colorectal cancer. What you should know. Oncology 20:579–587
Grotowski M (2002) Antigens (CEA and CA 19-9) in diagnosis and prognosis colorectal cancer. Pol Merkur Lekarski 12:77–80
Gygi SP, Corthals GL, Zhang Y et al (2000) Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc Natl Acad Sci USA 97:9390–9395
Haas BR, Sontheimer H (2010) Inhibition of the Sodium-Potassium-Chloride Cotransporter Isoform-1 reduces glioma invasion. Cancer Res 70:5597–5606
Hanash S (2003) Disease proteomics. Nature 422:226–232
Krausova M, Korinek V (2012) Signal transduction pathways participating in homeostasis and malignant transformation of the intestinal tissue. Neoplasma 59:708–718
Li N, Zhang J, Liang Y et al (2007) A controversial tumor marker: is SM22 a proper biomarker for gastric cancer cells? J Proteome Res 6:3304–3312
Li T, Yang W, Li M et al (2008) Expression of selenium-binding protein 1 characterizes intestinal cell maturation and predicts survival for patients with colorectal cancer. Mol Nutr Food Res 52:1289–1299
Melle C, Bogumil R, Ernst G et al (2006) Detection and identification of heat shock protein 10 as a biomarker in colorectal cancer by protein profiling. Proteomics 6:2600–2608
Murata T, Takayama K, Urano T et al (2012) 14-3-3zeta, a novel androgen-responsive gene, is upregulated in prostate cancer and promotes prostate cancer cell proliferation and survival. Clin Cancer Res 18:5617–5627
Nicolini A, Ferrari P, Duffy MJ et al (2010) Intensive risk-adjusted follow-up with the CEA, TPA, CA19.9, and CA72.4 tumor marker panel and abdominal ultrasonography to diagnose operable colorectal cancer recurrences: effect on survival. Arch Surg 145:1177–1183
Nuan J, Xin I, Jun W et al (2007) A proteomic method for analysis of CYP450 s protein expression changes in carbon tetrachloride induced male rat liver microsomes. Toxicology 237:1–11
Panagopoulos K, Cross-Knorr S, Dillard C et al (2013) Reversal of chemosensitivity and induction of cell malignancy of a non-malignant prostate cancer cell line upon extracellular vesicle exposure. Mol Cancer 12:118
Popa-Velea O, Cernat B, Tambu A (2010) Influence of personalized therapeutic approach on quality of life and psychiatric comorbidity in patients with advanced colonic cancer requiring palliative care. J Med Life 3:343–347
Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics. CA Cancer J Clin 64:9–29
Stulík J, Osterreicher J, Koupilová K et al (2000) Differential expression of the Ca2 + binding S100A6 protein in normal, preneoplastic and neoplastic colon mucosa. Eur J Cancer 36:1050–1059
Sun W, Gao S, Wang L et al (2006) Microwave assisted protein preparation and enzymatic digestion in proteomics. Mol Cell Proteom 5:769–776
Umar A, Luider TM, Foekens JA, Pasa-Tolić L (2007) NanoLC-FT-ICR MS improves proteome coverage attainable for approximately 3000 laser-microdissected breast carcinoma cells. Proteomics 7:323–329
Wang Z, Nesland JM, Suo Z et al (2011) The prognostic value of 14-3-3 isoforms in vulvar squamous cell carcinoma cases: 14-3-3beta and epsilon are independent prognostic factors for these tumors. PLoS ONE 6:e24843
Wheeler DB, Zoncu R, Root DE et al (2015) Identification of an oncogenic RAB protein. Science 350:211–217
Yonemori H, Kubota D, Taniguchi H et al (2013) Laser microdissection and two-dimensional difference gel electrophoresis with alkaline isoelectric point immobiline gel reveals proteomic intra-tumor heterogeneity in colorectal cancer. EuPA Open Proteom 1:17–29
Acknowledgments
We thank Xiaohong Qian and Xin Liu from Beijing Proteome Research Center for their technical assistance.
Funding
This study was supported by Beijing Postdoctoral Research Foundation. The funder played no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have stated that they have no conflicts of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, Y., Liu, Y., Ye, Y. et al. Quantitative proteome analysis of colorectal cancer-related differential proteins. J Cancer Res Clin Oncol 143, 233–241 (2017). https://doi.org/10.1007/s00432-016-2274-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-016-2274-5