
Abstract
Purpose
TNF-related apoptosis-inducing ligand (TRAIL) is a potential cancer therapeutic agent that preferentially induces apoptosis in cancer cells. However, breast cancer cells are generally resistant to TRAIL. Bufalin is a major active ingredient of the traditional Chinese medicine ChanSu. The present study aimed to assess the synergistic effect of bufalin and TRAIL and elucidate the underlying mechanisms in breast cancer cells.
Methods
Cell proliferation and apoptosis were measured by MTT assay and flow cytometry, respectively. The expression of proteins was assayed by flow cytometry and/or Western blotting. Transfection studies were used to determine the involvement of DR4, DR5 and Cbl-b in the synergistic effect of bufalin and TRAIL.
Results
MCF-7 and MDA-MB-231 cells were resistant to TRAIL. Both cell lines were dramatically sensitized to TRAIL-induced apoptosis by bufalin. Further experiments indicated that bufalin up-regulated DR4 and DR5, activated ERK, JNK and p38 MAPK and down-regulated Cbl-b. Blocking the up-regulation of DR4 and DR5 by siRNA rendered cells less sensitive to apoptosis induced by the combination of bufalin and TRAIL. Inhibition of the activation of ERK, JNK and p38 MAPK by specific inhibitors attenuated DR4 and DR5 up-regulation. Moreover, down-regulation of Cbl-b by shRNA led to stronger activation of ERK, JNK and p38 MAPK, more up-regulation of DR4 and DR5, and a stronger synergistic effect of bufalin and TRAIL.
Conclusions
Bufalin enhanced TRAIL-induced apoptosis by up-regulating the expression of DR4 and DR5. Bufalin-induced down-regulation of Cbl-b contributed to the up-regulation of DR4 and DR5, which might be partially mediated by the activation of ERK, JNK and p38 MAPK.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most common cause of death from cancer in women. A variety of cancer therapeutic methods, including surgery, radiation therapy, chemotherapy and endocrine therapy, have been utilized for the treatment of breast cancer. But more than 400,000 people still die from breast cancer each year in the world (Kamangar et al. 2006). Therefore, new treatment strategies are needed for this disease. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), a member of the tumor necrosis factor (TNF) superfamily, is a promising candidate for cancer therapy since it preferentially induces apoptosis in a variety of cancer cells with little or no effect on normal cells (Walczak et al. 1999; Roth et al. 1999). However, previous studies have shown that breast cancer cells are generally resistant to TRAIL-induced apoptosis (Keane et al. 1999). Hence, optimal TRAIL-based therapies for breast cancer will need to elucidate the mechanism of TRAIL resistance and incorporate agents to overcome the resistance. Several mechanisms have been proposed to explain resistance to TRAIL, including the expression levels of death receptors (DRs), FLICE inhibitor protein (FLIP) and Bcl-XL (Zhang and Fang 2005; Ashkenazi 2002). Previous studies have reported that chemotherapeutic drugs could sensitize cancer cells and xenograft tumors to TRAIL-induced apoptosis by increasing the expression of DR4 and/or DR5 (Kondo et al. 2006; Vondálová Blanárová et al. 2011). Recently, we reported that epirubicin could also enhance TRAIL-induced apoptosis by redistributing DR4 and DR5 in lipid rafts (Xu et al. 2011). Bufalin, a major active ingredient of the traditional Chinese medicine ChanSu, induces cell apoptosis or G2M cell cycle arrest in multiple types of cancer cells through altering the expression of Bcl-2/Bax, changing the intracellular concentration of Na+ or activation of c-jun NH2-terminal kinase (JNK) (Qi et al. 2011). Data from our laboratory and from other investigators have demonstrated that bufalin could enhance all-trans retinoic acid-induced differentiation in acute promyelocytic leukemia cells (Zhu et al. 2006; Yamada et al. 1998). Hashimoto et al. (1997) found that bufalin could also enhance the cytotoxicity of cisplatin and retinoic acid by reducing the level of topoisomeraseII in human leukemia cells. Recently, Dong et al. reported that bufalin could enhance TRAIL-induced apoptosis through down-regulating the expression of MCl-1 and BCL-XL (2011). However, whether bufalin could influence the expression of DR4 and DR5 in breast cancer is not clear.
In addition, recent studies have revealed that members of mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinase (ERK), JNK, and p38 MAPK, are important regulators in death receptor expression (Kang et al. 2011; Zou et al. 2004; Lepage et al. 2011). Our studies and those of others have shown that Casitas B-lineage lymphoma-b (Cbl-b), an E3 ubiquitin ligase and a multi-adaptor protein, could regulate apoptosis by controlling the activation of MAPKs and other signals (Sproul et al. 2009; Qu et al. 2004; Qu et al. 2009), and that c-Cbl, a homologue of Cbl-b, mediates the degradation of death receptors after TRAIL activation in human prostate adenocarcinoma cells (Song et al. 2010). However, there are no data regarding the ability of Cbl-b to regulate the expression of death receptors.
In the present study, we investigated the mechanism of the synergistic effect of bufalin and TRAIL in breast cancer cells. The results demonstrated that bufalin enhanced TRAIL-induced apoptosis via up-regulating the expression of DR4 and DR5. The bufalin-induced down-regulation of Cbl-b contributed to the up-regulation of death receptors, which might be partially mediated by the activation of ERK, JNK and p38 MAPK.
Materials and methods
Reagents and antibodies
Anti-β-actin (1:1,000 dilution), anti-ERK (1:2,000 dilution), anti-phospho-ERK (1:500 dilution) rabbit polyclonal antibodies, and anti-DR4 (1:500 dilution), anti-DR5 (1:500 dilution) mouse polyclonal antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phospho-JNK (1:250 dilution), anti-phospho-p38 MAPK (1:500 dilution), anti-JNK (1:500 dilution) and anti-PARP (1:1,000 dilution) rabbit polyclonal antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-Cbl-b (1:250 dilution) mouse polyclonal antibody was purchased from Transduction Laboratories (Lexington, KY, USA). Anti-caspase-8 (1:800 dilution) rabbit polyclonal antibody and anti-p38 MAPK (1:500 dilution) mouse polyclonal antibody were purchased from Neomarker (Fremont, CA, USA). Bufalin was purchased from Sigma–Aldrich (St. Louis, MO, USA). Recombinant human TRAIL was purchased from Cytolab/Peprotech Asia. The specific ERK inhibitor, PD98059, and the p38 MAPK inhibitor, SB203580, were purchased from Promega (Madison, WI, USA). The specific JNK inhibitor, SP600125, was purchased from Calbiochem (San Diego, CA, USA). The caspase-8 colorimetric assay kit was purchased from KeyGen Biotech. Co., Ltd (Nanjing, China).
Cell culture
The human breast cancer MCF-7 and MDA-MB-231 cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). MCF-7 cells were cultured in RPMI 1640 medium (GIBCO, Grand Island, NY). MDA-MB-231 cells were cultured in L15 medium (GIBCO, Grand Island, NY). Both RPMI 1640 and L15 medium were supplemented with 10 % heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were cultured at 37 °C under an atmosphere of 95 % air and 5 % CO2. Cells were routinely sub-cultured every 2–3 days, and the cell samples used were all in the logarithmic growth phase.
Cell viability assay
The effect of bufalin on MCF-7 and MDA-MB-231 cell proliferation was measured using the 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Cells were seeded at a density of 1 × 104 cells/well in 96-well plates and incubated overnight. Then, different concentrations of bufalin were added, and the cells were further incubated for the indicated times. Thereafter, 20 μl of the MTT solution (5 mg/ml) was added to each well, and the cells were incubated for another 4 h at 37 °C. After the removal of the culture medium, the cells were lysed in 200 μl of dimethylsulfoxide (DMSO), and then, the optical density (OD) was measured at 570 nm using a microplate reader (Bio-Rad, Hercules, CA, USA). The following formula was used: cell viability = (OD of the experimental sample/OD of the untreated group) × 100 %.
Flow cytometry assay
Phase distributions of the cell cycle and hypodiploid DNA were determined by flow cytometry. Cells were seeded at a density of 3.0 × 105 cells/well in 6-well plates and incubated overnight. After exposed to bufalin and/or TRAIL for the indicated times, the cells were collected and washed twice with phosphate-buffered saline (PBS). After fixing in ice-cold 70 % ethanol for 12 h, the samples were washed twice with PBS and then incubated with 20 μg/ml RNase A and 10 μg/ml propidium iodide (PI) for 30 min in the dark. The cell surface expression of DR4 and DR5 was performed by incubating the cells with specific primary antibody. Then, cells were stained with the secondary antibody conjugated with fluorescein isothiocyanate (FITC). Cells only stained with the secondary antibody were used as a negative control. Finally, the samples were evaluated by flow cytometry, and the data were analyzed with WinMDI software version 2.9 (The Scripps Research Institute, La Jolla, CA, USA).
Western blot analysis
Western blotting was performed using standard techniques as previously described. Briefly, cells were washed twice with ice-cold PBS and solubilized in 1 % Triton lysis buffer (1 % Triton X-100, 50 mM Tris–Cl pH 7.4, 150 mM NaCl, 10 mM EDTA, 100 mM NaF, 1 mM Na3VO4, 1 mM PMSF and 2 μg/ml aprotinin) on ice. The protein concentration was determined by the Lowry method. Total proteins (30–50 μg) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Immobilon-P, Millipore, Bedford, MA, USA). Membranes were blocked with 5 % skim milk in TBST (10 mM Tris, pH 7.4, 150 mM NaCl and 0.1 % Tween-20) at room temperature for 2 h and incubated with the indicated primary antibodies (see the “Materials” section) at 4 °C overnight. After washing with TBST, the membrane was reacted with the appropriate horseradish peroxidase–conjugated specific goat anti-mouse or goat anti-rabbit secondary antibodies (1:800 dilution) for 30 min at room temperature. After extensive washing with TBST, proteins were visualized by the enhanced chemiluminescence reagent (SuperSignal WesternPico Chemiluminescent Substrate; Pierce, Rockford, IL, USA).
Caspase activity assay
The enzymatic activity of caspase-8 was measured using a colorimetric assay kit (Keygen Biotech, Nanjing, China) following the manufacturer’s protocol. In brief, a total of 1.0 × 106 cells were seeded in the six wells and cultured for 24 h. Then, the cells were treated or untreated with TRAIL and bufalin for indicated times and harvested, resuspended in 50 μl of lysis buffer and incubated on ice for 60 min. The lysed cells were centrifuged at 10,000 rpm for 20 min, and equal amounts of protein were incubated with 50 μl of 2 × reaction buffer and 5 μl of peptide substrate, Ac-IETD-pNA for caspase-8, at 37 °C for 4 h in the dark. Samples were read at 405 nm in a microplate reader. Caspase-8 activity was calculated following the manufacturer’s instructions.
Reverse-transcription-polymerase chain reaction (RT-PCR)
MDA-MB-231 and MCF-7 cells either treated or untreated with bufalin were cultured and harvested at the indicated times. Cell pellets were washed twice with ice-cold PBS and total RNA extracted with the RNeasy mini kit (Qiagen, Carlsbad, CA, USA) as described by the manufacturer. RT-PCR was performed with primer pairs for DR4: forward (5′-CGATGTGGTCAGAGCTGGTACAGC-3′) and reverse (5′-GGACACGGCAGAGCCTGTGCCATC-3′), for DR5: forward (5′-GGGAGCCGCTCATGAGGAAGTTGG-3′) and reverse (5′-GGCAAGTCTCTCTCCCAGCGTCTC-3′), and for β-actin as a control: forward (5′-GTGGGG CGCCCCAGGCACCA-3′) and reverse (5′-CTCCTTAATGTCACGCACGATTTC-3′). PCR conditions were 95 °C for 5 min; 35 cycles of 95 °C for 45 s, 56 °C for 45 s, 72 °C for 45 s; one cycle of 72 °C for 10 min. The amplified products were then separated on 1.5 % agarose gels, stained with ethidium bromide and visualized under UV illumination.
Transfection with short hairpin RNA
One set of synthetic oligonucleotides encompassing the sense and antisense target sequences of human Cbl-b, 5′-GATCCCGTTTCCGGTTAAGTTGCACTCGTTCAAGAGACGAGTGCAACTTAACCGGAAATTTTTTCCAAA-3′ and 5′-AGCTTTTGGAAAAAATTTCCGGTTAAGTTGCACTCGTCTCTTGAACGAGTGCAACTTAACCGGAAAGG-3′ for Cbl-b (852), and one set of nonsilencing control, 5′-GATCCCGTTCTCCGAACGTGTCACGTTTGATATCCGACGTGACACGTTCGGAGAATTTTTTCCAAA-3′ and 5′-AGCTTTTGGAAAAAATTCTCCGAACGTGTCACGTCGGATACZAACGTGACACGTTCGGAGAA CGG-3′ were phosphorylated with T4 kinase (Takara, Tokyo, Japan), annealed and ligated into the Bam-HI/HindIII-cleaved backbone of pRNA-U6.1/Neo (Genscript, Piscataway, NJ, USA). Short hairpin RNA (ShRNA)-expressing vectors were transfected into MDA-MB-231 cells using Lipofectamine™ 2000 reagent (Invitrogen, Carlsbad, CA, USA). Stably transfected cells were screened with G418 (Invitrogen, Carlsbad, CA, USA). The expression of Cbl-b was verified by Western blotting. Three stably transfected cell lines were used for the following experiments.
Transfection with small interfering RNA
Cells were seeded at a density of 5 × 105 cells/well in 6-well plates. After 24 h, cells were transfected with small interfering RNA (siRNA) using Lipofectamine™ 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocols. DR4 and DR5 siRNA duplexes that target the sequences 5′-GGGTTACACCAATGCTTCCAACAAT-3′ and 5′-AAGACCCTTGTGCTCGTTGTC-3′, respectively, as described previously (Jin et al. 2005), were synthesized by Qiagen. Forty-eight hours after transfection, cells were treated with bufalin and TRAIL, and the controls were not treated with these agents. The gene silencing effect was evaluated by Western blotting.
Statistical analysis
All the presented data were confirmed in at least three independent experiments and are expressed as the mean ± SD. Statistical comparisons were made by Student’s t test. p < 0.05 was considered statistically significant. IC50 values were calculated by nonlinear regression analysis using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA, USA).
Results
Bufalin enhanced TRAIL-induced apoptosis in MCF-7 and MDA-MB-231 cells
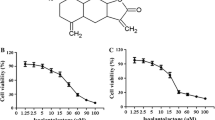
To evaluate the effect of bufalin on the proliferation of breast cancer cells, MCF-7 and MDA-MB-231 cells were treated with the indicated concentrations of bufalin for 24 h and 48 h. As shown by the MTT assay, bufalin triggered a time- and dose-dependent inhibition of proliferation (Fig. 1a, b). The 50 % inhibitory concentrations (IC50) at 24 and 48 h were 317.9 ± 1.5 nM and 46.5 ± 1.4 nM for MCF-7, 934.1 ± 2.0 nM and 513.3 ± 1.6 nM for MDA-MB-231 cells, respectively. Flow cytometry analysis showed that bufalin concentrations up to 50 nM induced apoptosis in 22.5 ± 2.5 % of MCF-7 cells at 48 h, but only in 6.5 ± 1.9 % of MDA-MB-231 cells (Fig. 1c). Western blot analysis showed that 50 nM of bufalin induced significant cleavage of poly (ADP-ribose) polymerase (PARP) at 24 h and at 48 h in MCF-7 cells, but only slight cleavage of PARP in MDA-MB-231 cells at 48 h (Fig. 1d). In the following experiment, we chose bufalin concentrations that could induce significant apoptosis in MCF-7 cells. To investigate the effect of bufalin on TRAIL-mediated apoptosis, MDA-MB-231 and MCF-7 cells were incubated with bufalin (50 nM) and/or TRAIL (100 ng/ml) for 24 h. Flow cytometry analysis showed that 100 ng/ml of TRAIL induced 2.0 ± 0.5 % apoptosis in MCF-7 cells, and 6.9 ± 1.8 % in MDA-MB-231 cells at 24 h (Fig. 1e), whereas our previous study showed that the same concentration of TRAIL induced >90 % apoptosis in TRAIL-sensitive Jurkat T cells (Liu et al. 2010). In the present study, bufalin increased TRAIL-induced apoptosis from 2.0 ± 0.5 % to 30.1 ± 1.2 % in MCF-7 cells, and from 6.9 ± 1.8 % to 41.5 ± 1.4 % in MDA-MB-231 cells (Fig. 1e). These results indicated that MCF-7 cells were more sensitive to bufalin than MDA-MB-231 cells. Both MCF-7 and MDA-MB-231 cells were resistant to TRAIL and were sensitized to TRAIL-induced apoptosis by bufalin.

Bufalin and TRAIL synergistically induced apoptosis in breast cancer cells. (a, b) MCF-7 and MDA-MB-231 cells were treated with different concentrations of bufalin for 24 h and 48 h. Cell growth inhibition was assessed by the MTT assay. Points represent means ± SD. Sigmoidal dose response curves were derived from fitting the data to a nonlinear regression program (Graph Pad Prism). c MCF-7 and MDA-MB-231 cells were treated with different concentrations of bufalin for 48 h. Apoptosis (APO) was analyzed as a sub-G1 fraction by flow cytometry with propidium iodide (PI) staining. The final results are summarized in the bar graphs. Data are means ± SD of three independent experiments. d MCF-7 and MDA-MB-231 cells were treated with 50 nM bufalin for the indicated times. PARP was detected by Western blotting. e MCF-7 and MDA-MB-231 cells were treated with 50 nM bufalin and/or 100 ng/ml TRAIL for 24 h. Apoptosis (APO) was analyzed as a sub-G1 fraction by flow cytometry with PI staining. The final results are summarized in the bar graphs. Data are means ± SD of three independent experiments (*p < 0.05 compared to the cells in the bufalin treated group; # p < 0.05 compared to the cells in the untreated group)
Up-regulation of DR4 and DR5 was involved in the synergistic effect of bufalin and TRAIL
To assess the molecular mechanisms underlying the synergistic induction of apoptosis by bufalin and TRAIL in breast cancer cells, the activation of caspase-8 and the cleavage of PARP were measured. As shown in Fig. 2a and b, TRAIL alone failed to induce the cleavage of caspase-8. Combining bufalin with TRAIL enhanced cell apoptosis, which was accompanied by increasing the cleavage of PARP, enhancing the activation of procaspase-8, in both MCF-7 and MDA-MB-231 cells. These results indicated that bufalin sensitized both MDA-MB-231 and MCF-7 breast cancer cells to TRAIL-induced apoptosis by activating the extrinsic apoptotic pathway. Next, we examined the effect of bufalin on the expression of DR4 and DR5. Western blot analysis showed that the expression of DR4 and DR5 was significantly up-regulated after bufalin treatment in both MCF-7 and MDA-MB-231 cells (Fig. 2c, d). The up-regulation of DR4 and DR5 was further confirmed by flow cytometric analysis using specific antibodies. Flow cytometric analysis showed that the expression levels of DR4 and DR5 were markedly higher in cells treated with bufalin than the untreated control cells (Fig. 2e). RT-PCR analysis showed up-regulation of DR4 and DR5 mRNA after treatment with bufalin that correlated with the increase of DR4 and DR5 proteins (Fig. 2f). We next examined whether the up-regulation of DR4 and DR5 is directly associated with bufalin + TRAIL-induced apoptosis, employing siRNA duplexes against DR4 or DR5 mRNA. MDA-MB-231 cells transfected with the control siRNA, DR4 siRNA and/or DR5 siRNA were either untreated or treated with 50 nM of bufalin and 100 ng/ml of TRAIL for 24 h. Flow cytometric analysis showed that blocking of DR4 and DR5 expression alone significantly reduced the rate of cell death. Cotransfection of DR4 and DR5 siRNAs together was more potent than either siRNA alone in decreasing bufalin and TRAIL-induced apoptosis (Fig. 2g). Taken together, these results suggested that up-regulation of DR4 and DR5, at least in part, directly contributed to the synergistic effect of bufalin and TRAIL.

Bufalin enhanced TRAIL-induced apoptosis by activating the extrinsic apoptotic pathway and up-regulating DR4 and DR5. (a, b) MCF-7 and MDA-MB-231 cells were untreated, treated with 50 nM bufalin and/or 100 ng/ml TRAIL for 24 h. The PARP and caspase-8 were analyzed by Western blotting. (c, d) MCF-7 and MDA-MB-231 cells were untreated, or treated with 50 nM bufalin for indicated times. The expression of DR4 and DR5 was analyzed by Western blotting. (e) MCF-7 and MDA-MB-231 cells were treated with 50 nM of bufalin for 24 h, and then, DR4 and DR5 were detected by flow cytometry. The filled red peaks represent cells stained with the FITC-conjugated secondary antibody only. The open peaks are cells stained with primary antibody against an individual death receptor followed by the FITC-conjugated secondary antibody. f MDA-MB-231 and MCF-7 cells were treated with 50 nM bufalin for indicated times, and the expression of mRNA for DR4 and DR5 was analyzed by RT-PCR. β-actin was used as an internal control. M indicated marker. g MDA-MB-231 cells were transiently transfected with control (Ctrl), DR4 and/or DR5 siRNA. At 48 h after transfection, the cells were treated with the combination of 50 nM bufalin and 100 ng/ml TRAIL (Bufalin/TRAIL) for 24 h. The expression of DR4 and DR5 proteins was analyzed by Western blotting. Apoptosis was analyzed as a sub-G1 fraction by flow cytometry with PI staining. The final results are summarized in the bar graphs. Data are means ± SD of three independent experiments (*p < 0.05 compared to the cells in the Ctrl siRNA transfected group)
Activation of MAPK signaling pathways contributed to the up-regulation of DR4 and DR5
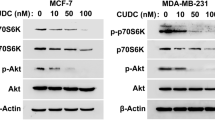
Recent studies have revealed that MAPKs are important regulators of death receptor expression. To investigate the role of MAPKs in bufalin-induced DR4 and DR5 expression, Western blot analysis was used to assess the change in phosphorylations of ERK, JNK and p38 MAPK. The results showed that ERK, JNK and p38 MAPK were activated by bufalin in MDA-MB-231 cells (Fig. 3a). Thus, MDA-MB-231 cells were pretreated with the ERK inhibitor PD98059, the p38 MAPK inhibitor SB239063 or the JNK inhibitor SP600125 and then incubated with bufalin to investigate the functional roles of these activations. The results showed that inhibition of ERK, JNK and p38 MAPK all restored the apoptosis induced by treatment with bufalin and TRAIL (Fig. 3b, c). We also investigated whether MAPKs activation contributed directly to the up-regulation of DR4 and DR5. Consistent with the decrease in apoptosis, pre-treatment with PD98059, SB239063 or SP600125 decreased bufalin-induced DR4 and DR5 up-regulation, even though p38 MAPK had relative moderate effect on DR4 expression (Fig. 3d). These data indicated that the MAPK signal pathways were regulators of bufalin-induced DR4 and DR5 expression. The up-regulation of DR4 and DR5 played an important role during bufalin + TRAIL-induced apoptosis.

The role of MAPK signaling pathways in bufalin-induced up-regulation of DR4 and DR5. a MDA-MB-231 cells were treated with 50 nM bufalin for the indicated times. The activities of ERK, JNK and p38 MAPK were analyzed by Western blotting. b MDA-MB-231 cells were pretreated with 10 μM SB239063 (SB), 10 μM SP600125 (SP) or 20 μM PD98059 (PD) for 2.5 h and then treated with 50 nM bufalin and/or 100 ng/ml TRAIL for 24 h. Apoptosis (APO) was analyzed as a sub-G1 fraction by flow cytometry with PI staining. The final results are summarized in the bar graphs. c Data are means ± SD of three independent experiments (*p < 0.05 compared to the cells in the bufalin + TRAIL-treated group; # p < 0.05 compared to the cells in the TRAIL-treated group). d MDA-MB-231 cells were pretreated with 10 μM SB239063 (SB), 10 μM SP600125 (SP) or 20 μM PD98059 (PD) for 2.5 h and then treated with 50 nM bufalin for 24 h. The expression of DR4 and DR5 was analyzed by Western blotting
Bufalin up-regulated the expression of DR4 and DR5 by down-regulating the expression of Cbl-b
To investigate the mechanism of bufalin-induced up-regulation of DR4 and DR5, we detected the expression of c-Cbl and Cbl-b in MCF-7 and MDA-MB-231 cells after bufalin treatment. Western blot analysis showed that the expression of c-Cbl was not influenced by bufalin treatment, but Cbl-b was down-regulated after bufalin treatment in a time-dependent manner in both MCF-7 and MDA-MB-231 cells (Fig. 4a). To test whether targeting Cbl-b was the effective strategy by which bufalin enhanced TRAIL-induced apoptosis, the expression of Cbl-b was down-regulated in MDA-MB-231 cells by shRNA. Flow cytometry analysis showed that Cbl-b shRNA-transfected (Cbl-b.shRNA) cells were more sensitive to bufalin + TRAIL-induced apoptosis than the control shRNA-transfected (Ctrl.shRNA) cells (Fig. 4b, c). Western blot analysis showed that apoptosis of a higher number of cells was accompanied by more cleavage of PARP, and stronger activation of procaspase-8 after bufalin + TRAIL treatment in Cbl-b.shRNA cells than Ctrl.shRNA cells. At the same time, DR4 and DR5 were up-regulated more significantly in Cbl-b.shRNA cells than in Ctrl.shRNA cells after bufalin and bufalin + TRAIL treatments. Endogenous DR4 and DR5 were up-regulated slightly in Cbl-b.shRNA cells, which was consistent with the flow cytometry analysis indicating that the Cbl-b.shRNA cells were slightly more sensitive to TRAIL than Ctrl.shRNA cells (Fig. 4d). The activity of caspase-8 was further confirmed using a colorimetric assay kit (Fig. 4e). These results indicated that the down-regulation of Cbl-b contributed to bufalin-induced up-regulation of DR4 and DR5.

Cbl-b was involved in regulating the synergistic effect of bufalin and TRAIL by regulating the expression of DR4 and DR5. a MCF-7 and MDA-MB-231 cells were treated with 50 nM bufalin for the indicated times. The expression of c-Cbl and Cbl-b was analyzed by Western blotting. b MDA-MB-231 cells were stably transfected with control shRNA or Cbl-b shRNA. The control shRNA-transfected cells (Ctrl.shRNA) and Cbl-b shRNA-transfected cells (Cbl-b.shRNA) were untreated or treated with 50 nM bufalin and/or 100 ng/ml TRAIL for 24 h. Apoptosis (APO) was analyzed as a sub-G1 fraction by flow cytometry with PI staining. The final results are summarized in the bar graphs. c Data are means ± SD of three independent experiments (*p < 0.05 compared to the cells in the Ctrl.shRNA group). d The Ctrl.shRNA and Cbl-b.shRNA cells were treated with 50 nM bufalin and/or 100 ng/ml TRAIL for 16 h. The expression of the indicated proteins was analyzed by Western blotting. e The Ctrl.shRNA and Cbl-b.shRNA cells were treated or untreated with 50 nM bufalin and 100 ng/ml TRAIL for 16 h. The activity of caspase-8 was assessed using a colorimetric assay kit. Data are given as the fold increase above control. Data are means ± SD of three independent experiments (*p < 0.05 compared to the cells in the Ctrl.shRNA group)
The relationship between the down-regulation of Cbl-b and the activation of MAPK signaling pathways
To explore the relationship between the down-regulation of Cbl-b and the activation of MAPK signaling pathways, the activation of MAPKs was assessed after knockdown of Cbl-b. As shown in Fig. 5, bufalin and bufalin + TRAIL-induced activation of ERK, JNK and p38 MAPK were significantly increased after down-regulation of Cbl-b by shRNA. Endogenous activation of MAPKs was slightly enhanced after Cbl-b downregulation. These results implied that Cbl-b might be involved in the regulation of MAPKs activation induced by bufalin.

The relationship between Cbl-b down-regulation and MAPKs activation. MDA-MB-231 cells were stably transfected with control shRNA or Cbl-b shRNA. The control (Ctrl.shRNA) and Cbl-b shRNA-transfected (Cbl-b.shRNA) cells were treated with 50 nM bufalin and/or 100 ng/ml TRAIL for 16 h. The activation of p38 MAPK, JNK and ERK was analyzed by Western blotting
Discussion
TRAIL, a potential anti-cancer biological agent, is currently under a phase II clinical trial (Bellail et al. 2009; Duiker et al. 2006). However, resistance to TRAIL has been a major obstacle for clinical administration. Therefore, it is important to develop novel enhancers to overcome TRAIL resistance. The present study revealed that breast cancer MCF-7 and MDA-MB-231 cells were resistant to TRAIL, in contrast to human acute leukemia Jurkat cells (Liu et al. 2010). Both MCF-7 and MDA-MB-231 cells were sensitized to TRAIL by bufalin, although the two cell lines had different sensitivities to bufalin. This observation indicates that bufalin is a potent enhancer of TRAIL-mediated apoptosis, and this effect warrants additional clinical studies.
Binding of TRAIL to DR4 and DR5 leads to caspase-8-initiated extrinsic pathway activation and subsequent cleavage of cellular substrates, such as PARP, leading to cell apoptosis (MacFarlane 2003). The expression of DR4 and/or DR5 plays a crucial role in synergistic cytotoxicity associated with TRAIL and chemotherapeutic drugs (Srivastava 2001; Ashkenazi and Dixit 1999; Gómez-Benito et al. 2007). Recently, Dong et al. reported that bufalin could sensitize breast cancer cells to TRAIL by down-regulating the expression of MCl-1 and BCL-XL (2011). However, there are no data on whether the change of DR4 and DR5 is involved in this process. In the present study, bufalin enhanced TRAIL-induced apoptosis, which was accompanied by the up-regulation of DR4 and DR5. Blocking the up-regulation of the receptors by siRNA against DR4 and DR5 attenuated the apoptosis, suggesting that the up-regulation of DR4 and DR5 was involved in the synergistic effect of bufalin and TRAIL. It has been reported that the expression of death receptors is regulated through p53-dependent and p53-independent mechanisms under some stress, or after treatment with chemotherapeutic drugs (Wang and El-Deiry 2003; Sheikh and Fornace 2000). Recently, c-Cbl was reported to be a down-regulator of TRAIL receptors after TRAIL treatment in human prostate adenocarcinoma DU-145 cells (Song et al. 2010). The present study revealed that Cbl-b, a homologue of c-Cbl, was down-regulated by bufalin. Down-regulation of Cbl-b by shRNA enhanced the synergistic effect of bufalin and TRAIL, along with more up-regulation of DR4 and DR5 after bufalin and bufalin + TRAIL treatment. These results demonstrate that Cbl-b is a regulator of cell sensitivity to the combination of bufalin and TRAIL. In addition, the results also suggest for the first time that Cbl-b is a negative regulator of agent-induced DR4 and DR5 expression.
Recently, it has been reported that ERK, JNK and p38 MAPK are involved in agent-induced DR5 up-regulation in human leukemia THP-1 cells, human lung cancer cells and colon cancer cells, respectively (Kang et al. 2011; Zou et al. 2004; Lepage et al. 2011). The present study showed that ERK, JNK and p38 MAPK were activated by bufalin, which paralleled the inducible expression of DR4 and DR5. Inhibition of ERK, JNK or p38 MAPK attenuated bufalin-induced DR4 and DR5 up-regulation and bufalin + TRAIL-induced apoptosis. These observations suggest that the three signaling molecules are involved in bufalin-induced up-regulation of DR4 and DR5 in breast cancer cells. Sproul et al. have reported that in response to apoptotic stimuli, Cbl-b levels fall rapidly, relieving its inhibition of apoptotic JNK signaling in healthy neurons (2009). Cbl-b overexpression strongly inhibits ERK activation in mast cells (Qu et al. 2004). Sohn et al. have reported that Cbl-b silencing results in increases in ERK and JNK phosphorylation and surface expression of CD69 in mature B cells (2003). Although there are no data on whether Cbl-b could regulate the activation of the p38 MAPK signaling pathway, we speculate that down-regulation of Cbl-b might up-regulate the expression of DR4 and DR5 by disinhibiting the activation of MAPKs. Consistent with the speculation, our results showed that down-regulation of Cbl-b by shRNA resulted in more significant activation of ERK, JNK and p38 MAPK after bufalin and bufalin + TRAIL treatment. These results suggest that the down-regulation of Cbl-b results in the up-regulation of DR4 and DR5, which might be partially mediated by the activation of MAPKs. However, inhibition of Cbl-b expression resulted in slight activation of the endogenous MAPKs and sequential slight upregulation of endogenous DR4/DR5 expression. Accordingly, TRAIL treatment as a single agent only slightly increased the cell death in the Cbl-b-depleted cells, compared with the control cells.
Moreover, the results demonstrate for the first time that Cbl-b was a negative regulator of the p38 MAPK signaling pathway. The results showed that p38 MAPK had a more pronounced effect on bufalin + TRAIL-induced apoptosis, although it had a relatively moderate effect on DR4 expression. The reason for this inconsistency might be that p38 MAPK regulated bufalin + TRAIL-induced apoptosis through a complicated mechanism besides regulating the expression of DR4 and DR5, such as by inhibiting the activity of Stat3 (Ahmed et al. 2002), a target for p38 MAPK, that regulates bufalin + TRAIL-induced apoptosis through up-regulating Mcl-1 and BCL-XL (Dong et al. 2011).
In summary, we have demonstrated in the present study that bufalin potentiates the apoptotic effect of TRAIL in human breast cancer cells in part by regulating the expression of DR4 and DR5. The down-regulation of Cbl-b results in up-regulation of DR4 and DR5, which might be partially mediated by the activation of MAPKs. These results provide insight into the design of future combination therapies for cancer treatment with TRAIL. The mechanism for the regulation of Cbl-b expression by bufalin is currently being investigated in our laboratory.
References
Ahmed ST, Mayer A, Ji JD et al (2002) Inhibition of IL-6 signaling by a p38-dependent pathway occurs in the absence of new protein synthesis. J Leukoc Biol 72:154–162
Ashkenazi A (2002) Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer 2:420–430
Ashkenazi A, Dixit VM (1999) Apoptosis control by death and decoy receptors. Curr Opin Cell Biol 11:255–260
Bellail AC, Qi L, Mulligan P et al (2009) TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev Recent Clin Trials 4:34–41
Dong Y, Yin S, Li J et al (2011) Bufadienolide compounds sensitize human breast cancer cells to TRAIL-induced apoptosis via inhibition of STAT3/Mcl-1 pathway. Apoptosis 16:394–403
Duiker EW, Mom CH, de Jong S et al (2006) The clinical trail of TRAIL. Eur J Cancer 42:2233–2240
Gómez-Benito M, Martinez-Lorenzo MJ, Anel A et al (2007) Membrane expression of DR4, DR5 and caspase-8 levels, but not Mcl-1, determine sensitivity of human myeloma cells to Apo2L/TRAIL. Exp Cell Res 313:2378–2388
Hashimoto S, Jing Y, Kawazoe N et al (1997) Bufalin reduces the level of topoisomerase II in human leukemia cells and affects the cytotoxicity of anticancer drugs. Leuk Res 21:875–883
Jin F, Liu X, Zhou Z et al (2005) Activation of nuclear factor-kappaB contributes to induction of death receptors and apoptosis by the synthetic retinoid CD437 in DU145 human prostate cancer cells. Cancer Res 65:6354–6363
Kamangar F, Dores GM, Anderson WF (2006) Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol 24:2137–2150
Kang CH, Moon DO, Choi YH et al (2011) Piceatannol enhances TRAIL-induced apoptosis in human leukemia THP-1 cells through Sp1- and ERK-dependent DR5 up-regulation. Toxicol In Vitro 25:605–612
Keane MM, Ettenberg SA, Nau MM et al (1999) Chemotherapy augments TRAIL-induced apoptosis in breast cell lines. Cancer Res 59:734–741
Kondo K, Yamasaki S, Sugie T et al (2006) Cisplatin-dependent up-regulation of death receptors 4 and 5 augments induction of apoptosis by TNF-related apoptosis-inducing ligand against esophageal squamous cell carcinoma. Int J Cancer 118:230–242
Lepage C, Léger DY, Bertrand J et al (2011) Diosgenin induces death receptor-5 through activation of p38 pathway and promotes TRAIL-induced apoptosis in colon cancer cells. Cancer Lett 301:193–202
Liu J, Qu X, Xu L et al (2010) Phosphoinositide 3-kinase/Akt and nuclear factor κB pathways are involved in tumor necrosis factor-related apoptosis-inducing ligand resistance in human gastric cancer cells. Mol Med Report 3:491–496
MacFarlane M (2003) TRAIL-induced signalling and apoptosis. Toxicol Lett 2003(139):89–97
Qi F, Li A, Inagaki Y et al (2011) Antitumor activity of extracts and compounds from the skin of the toad Bufo bufo gargarizans Cantor. Int Immunopharmacol 11:342–349
Qu X, Sada K, Kyo S et al (2004) Negative regulation of FcepsilonRI-mediated mast cell activation by a ubiquitin-protein ligase Cbl-b. Blood 103:1779–1786
Qu X, Zhang Y, Li Y et al (2009) Ubiquitin ligase Cbl-b sensitizes leukemia and gastric cancer cells to anthracyclines by activating the mitochondrial pathway and modulating Akt and ERK survival signals. FEBS Lett 583:2255–2262
Roth W, Isenmann S, Naumann U et al (1999) Locoregional Apo2L/TRAIL eradicates intracranial human malignant glioma xenografts in athymic mice in the absence of neurotoxicity. Biochem Biophys Res Commun 265:479–483
Sheikh MS, Fornace AJ Jr (2000) Death and decoy receptors and p53-mediated apoptosis. Leukemia 14:1509–1513
Sohn HW, Gu H, Pierce SK (2003) Cbl-b negatively regulates B cell antigen receptor signaling in mature B cells through ubiquitination of the tyrosine kinase Syk. J Exp Med 197:1511–1524
Song JJ, Szczepanski MJ, Kim SY et al (2010) c-Cbl-mediated degradation of TRAIL receptors is responsible for the development of the early phase of TRAIL resistance. Cell Signal 22:553–563
Sproul AA, Xu Z, Wilhelm M et al (2009) Cbl negatively regulates JNK activation and cell death. Cell Res 19:950–961
Srivastava RK (2001) TRAIL/Apo-2L: mechanisms and clinical applications in cancer. Neoplasia 3:535–546
Vondálová Blanárová O, Jelínková I, Szöor A et al (2011) Cisplatin and a potent platinum(IV) complex-mediated enhancement of TRAIL-induced cancer cells killing is associated with modulation of upstream events in the extrinsic apoptotic pathway. Carcinogenesis 32:42–51
Walczak H, Miller RE, Ariail K et al (1999) Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 5:157–163
Wang S, El-Deiry WS (2003) TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 22:8628–8633
Xu L, Qu X, Luo Y et al (2011) Epirubicin enhances TRAIL-induced apoptosis in gastric cancer cells by promoting death receptor clustering in lipid rafts. Mol Med Report 4:407–411
Yamada K, Hino K, Tomoyasu S et al (1998) Enhancement by bufalin of retinoic acid-induced differentiation of acute promyelocytic leukemia cells in primary culture. Leuk Res 22:589–595
Zhang L, Fang B (2005) Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther 12:228–237
Zhu ZT, Jin B, Liu YP et al (2006) Enhancement of all-trans retinoic acid-induced differentiation by bufalin in primary culture of acute promyelocytic leukaemia cells. Zhonghua Nei Ke Za Zhi 45:314–317
Zou W, Liu X, Yue P et al (2004) c-Jun NH2-terminal kinase-mediated up-regulation of death receptor 5 contributes to induction of apoptosis by the novel synthetic triterpenoid methyl-2-cyano-3,12-dioxooleana-1, 9-dien-28-oate in human lung cancer cells. Cancer Res 64:7570–7578
Acknowledgements
This work was supported by grants from the National Science Foundation of China (No. 81172369, No. 81172198 and No.81172535).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yan, S., Qu, X., Xu, C. et al. Down-regulation of Cbl-b by bufalin results in up-regulation of DR4/DR5 and sensitization of TRAIL-induced apoptosis in breast cancer cells. J Cancer Res Clin Oncol 138, 1279–1289 (2012). https://doi.org/10.1007/s00432-012-1204-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-012-1204-4