
Abstract
Purpose
Docetaxel plus capecitabine, a commonly used chemotherapeutic regimen for metastatic breast cancer (MBC), is highly variable in its effectiveness. We aimed to investigate whether allelic variants of cytochrome P450 (CYP450) affected objective response, progression-free survival (PFS), and overall survival (OS) in MBC.
Patients and methods
79 SNPs in CYP450, whose minor allele frequency were ≥10%, were genotyped in 69 MBC patients who were treated with docetaxel plus capecitabine. Pearson’s χ2 test or Fisher’s exact test was used to investigate the influence of SNPs on objective response as appropriate. Log-rank test was used to assess the association between SNPs and survival outcomes.
Results
There is no significant association between polymorphisms and both objective response and OS. Only one SNP, CYP1A1 rs1048943 A>G (Ile462Val), was significantly associated with PFS (P = 0.0003). Multivariate analysis confirmed its prognostic significance for PFS (P = 0.004).
Conclusion
CYP1A1 rs1048943 A>G (Ile462Val) polymorphism is a potential prognostic marker for survival outcome after docetaxel plus capecitabine chemotherapy in MBC patients. However, confirmatory study is needed to validate this finding.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most frequently diagnosed cancer and the leading cause of cancer-related deaths in females worldwide, accounting for about 1.4 million new cases and 0.5 million deaths annually (Jemal et al. 2011). Due a large part to improvements in screening detection, more and more patients are diagnosed at an early stage. Unfortunately, about 1/3 of them eventually develop metastatic disease (Harbeck et al. 2010; Moreno-Aspitia and Perez 2009), the prognosis for whom remains poor, with a median survival of 2–3 years and 5-year survival of 26.7% (Moreno-Aspitia and Perez 2009).
Chemotherapy is an important therapeutic option for metastatic breast cancer (MBC), with anthracyclines and taxanes the two most active kinds of agents. The increasing use of anthracyclines and taxanes in adjuvant and neoadjuvant settings has led to a growing number of MBC who relapsed after treatment with these agents. For patients in whom anthracyclines have failed, docetaxel plus capecitabine are effective as demonstrated in previous studies (O’Shaughnessy et al. 2002; Chan et al. 2009; Michalaki et al. 2009). However, objective response can only be observed in about 40% of patients, while the remaining patients display various levels of resistance (O’Shaughnessy et al. 2002; Chan et al. 2009; Michalaki et al. 2009). Despite great efforts and major funds to identify appropriate predictive marker, we still cannot definitely distinguish responders from nonresponders before drug administration. Although factors such as poor compliance, pathophysiological conditions, and drug–drug interactions might affect the therapeutic outcome, gene constitution may account for 20–95% of the variability in drug effects (Ingelman-Sundberg et al. 2007; Watson and McLeod 2011).
Pharmacogenetic variation can be envisioned in four levels: drug metabolizing enzymes, drug transporters, drug targets, and other biomarker genes, with drug metabolizing enzymes the most prominent one. Genetic polymorphisms of drug transporters and drug receptors do, in some cases, determine therapeutic outcome, but the number of important examples where this variation is of clinical importance are relatively few (Ingelman-Sundberg et al. 2007). Whereas genetic polymorphisms in drug metabolizing enzymes can affect 20–25% of all drug therapies to an extent that therapy outcome is altered (Ingelman-Sundberg 2004), and cytochrome P450 (CYP450) plays a critical role because it involves in about 80% of all phase I drug metabolisms. Furthermore, genetic polymorphisms of CYP450 have been reported to be associated with breast cancer risk (Ma et al. 2010; Sergentanis and Economopoulos 2010), differentiation (Justenhoven et al. 2010), clinical outcome of hormonal therapy (Kiyotani et al. 2010; Ruiter et al. 2010), and response and survival after some regimens of chemotherapy (Bray et al. 2010; Gor et al. 2010; Petros et al. 2005). But to the best of our knowledge, there is no evidence whether genetic polymorphisms in CYP450 can affect clinical outcomes of a commonly used chemotherapeutic regimen of docetaxel plus capecitabine in patients with MBC.
In this study, we evaluated the impact of the single nucleotide polymorphisms (SNP) of CPY450 on objective response, progression-free survival, and overall survival, with the aim of identifying predictive and prognostic factors.
Patients and methods
Study population
This was a single-institute, prospective study. Patients with MBC who had been previously treated with anthracycline were included into our study in Breast Oncology Department of Peking University Cancer Hospital and Institute from January 2007 to June 2010. Eligibility criteria also included female, aged ≥18 years, Eastern Cooperative Oncology Group (ECOG) performance status of 0–2, adequate cardiac (normal left ventricular ejection fraction [LVEF > 50%]), hepatic (serum transaminases ≤1.5 × upper limit of normal [ULN], alkaline phosphatase ≤2.5 × ULN, bilirubin ≤1.0 × ULN), renal (serum creatinine ≤1.5 × ULN), and bone marrow (absolute neutrophil count ≥1.5 × 109/L, platelet count ≥100 × 109/L, hemoglobin ≥9 g/dL) function, at least one measurable lesion according to the response evaluation criteria in solid tumors (RECIST) version 1.0 (Therasse et al. 2000), and life expectancy of at least 3 months. Prior hormonal therapy, radiotherapy or chemotherapy were all allowed if they had been completed at least 4 weeks before enrollment and if <25% of the active bone marrow had been irradiated.
Exclusion criteria included previous exposure to docetaxel or capecitabine, active central nervous system metastasis, active infection, serious concurrent medical illness, pregnancy or lactation, previous radiotherapy on the assessable lesion, and history of another malignancy (except basal cell skin carcinoma and carcinoma in situ of the uterine cervix) within 5 years of study entry. Concomitant radiotherapy or hormone therapy was not permitted.
Patients received docetaxel (75 mg/m2, d1) and capecitabine (950 mg/m2 twice daily, on days 1–14) based on a triweekly cycle. Treatment interruption or dose reduction was made on the basis of hematologic and nonhematological toxicities. Treatment was interrupted in cases of grade 2 or higher toxicities (with the exception of toxicities considered by the investigator unlikely to become serious or life threatening, e.g., alopecia) and was not resumed until the adverse event was improved to grade 1 or 0. The doses of both drugs were reduced by 25% for patients who experienced a second occurrence of a given grade 2 toxicity or any grade 3 toxicity. Docetaxel therapy was discontinued if toxicities did not resolve to grade 0 or 1 within 2 weeks, but capecitabine could be resumed at 75% of the starting dose on resolution of the toxicity to grade 0 or 1. If patients experienced a third occurrence of a given grade 2 toxicity, a second occurrence of a given grade 3 toxicity, or any grade 4 toxicity, the capecitabine dose was reduced by 50% and docetaxel was discontinued. Capecitabine was discontinued if, despite dose reduction, a given toxicity occurred for a fourth time at grade 2, a third time at grade 3, or a second time at grade 4.
Chemotherapy was stopped in case of disease progression, patient refusal, or unacceptable toxicity. Tumor response was measured every two cycles by computed tomography scan to document complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD) according to RECIST guidelines version 1.0 (Therasse et al. 2000). Adverse events were classified according to National Cancer Institute Common Toxicity Criteria (NCI-CTC) version 3.0. The study was approved by the Ethics Committee of Peking University Cancer Hospital and Institute. Written informed consent for genetic polymorphism analysis was obtained from all patients before enrollment, and the study was carried out in accordance with Declaration of Helsinki.
Genotyping of CYP450 gene polymorphisms
Genomic DNA was extracted from venous blood samples (4 ml) drawn before drug administration, using standard phenol–chloroform method. The selection of SNPs is based on the Affymetrix Drug Metabolizing Enzymes and Transporters (DMET) genotyping platform, which integrates 1,936 genetic variations in 225 genes associated with phase I–II drug metabolism, disposition, and transport. Specifically, DMET platform included 438 genetic variations in CYP450. Only 79 SNPs of the 438 genetic variations have minor allele frequency (MAF) of ≥10% in Chinese based on HapMap Project database (http://www.hapmap.org). All the 79 SNPs were included into our study. These genes, reference SNP identification numbers, nucleotide substitutions, functions, chromosome position, and reported allele frequencies were summarized in supplementary data Table S1. 66 SNPs were genotyped by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry method (Sequenom, Inc., San Diego) and 13 by polymerase chain reaction (PCR) and direct sequencing (supplementary data Table S1). To verify the results, 5% of the DNA samples were randomly selected for duplicate assays. All the results were generated and analyzed by laboratory staff unaware of patient status.
Statistical analysis
Treatment response outcomes were dichotomized by responders (CR + PR) and nonresponders (SD + PD). PFS was calculated from the date therapy was initiated to the date of progression or death. OS was measured from the date therapy was initiated to the date of death. Patients who were alive were censored at the date of the last follow-up. Median PFS and OS were estimated according to the Kaplan–Meier method.
First, we analyzed the effects of demographic and clinical parameters on objective response, PFS, and OS. The demographic and clinical parameters included age (≤median vs. >median), ECOG performance status (0 vs. ≥1), tumor grading (I vs. II vs.III), estrogen receptor (negative vs. positive), progestin receptor (negative vs. positive), human epidermal growth factor receptor-2 (negative vs. positive), menopausal status (premenopause vs. postmenopause), treatment-line (first-line vs. ≥second-line), previous exposure to paclitaxel (yes vs. no), and visceral metastasis (yes vs. no). Pearson’s χ2 test or Fisher’s exact test was used to investigate the influence of demographic and clinical parameters on objective response as appropriate, and Log-rank test on PFS and OS.
Genotype distributions were checked for agreement with those expected under Hardy–Weinberg equilibrium using the goodness-of-fit χ2 test. Pearson’s χ2 test or Fisher’s exact test was used to investigate the influence of SNPs on objective response as appropriate. Log-rank test was used to assess the effects of SNPs on PFS and OS. In the univariate analyses for clinical outcome according to the 79 studied polymorphisms, a Bonferroni correction required a P < 0.05/79 = 0.0006 for statistical significance.
Cox proportional hazards multivariate modeling was used to identify factors that independently predict PFS and OS. All data were analyzed by SPSS for windows version 16.0 (SPSS Inc., Chicago, IL, USA). A cutoff P value of 0.05 was adopted for all the statistical analysis, except for the association between SNPs and clinical outcomes, which used 0.0006 for statistical significance.
Results
Patient characteristics
A total of 69 patients were enrolled into the current study. The median age was 55 (range, 29–75) years. Their baseline characteristics were shown in Table 1. Most patients were ECOG performance status of 0 (52.2%), postmenopausal (73.9%), previously treated by paclitaxel (69.6%), and received docetaxel plus capecitabine in the first-line setting (69.6%). Estrogen receptor, progestin receptor, and HER-2 over expressing status were examined on the tissue of the primary tumors in 48 patients, on metastatic lesion in 20 patients, and unknown in 1 patient.
Totally, 277 treatment cycles (median 4, range 1–8) were delivered. One patient received only one cycle of chemotherapy and could not be evaluated for response. Among 68 evaluable patients, the response rate was 27.9%, with no complete response, 19 partial responses (27.9%), 27 stable diseases (39.7%), and 22 progressive diseases (32.4%). The median PFS and OS was 6.0 months (95% CI, 5.3–6.7) and 17.1 months (95% CI, 13.0–21.2), respectively.
Effects of demographic and clinical variables on objective response, PFS, and OS
As shown in supplementary data Table S2, none of the demographic and clinical variables was statistically related to objective response, while positive estrogen receptor and postmenopausal status were associated with longer PFS (P = 0.034 and P = 0.040, respectively, as shown in Fig. 1), and ECOG performance status of 0 and postmenopausal status were associated with longer OS (P = 0.001 and P = 0.023, respectively, as shown in Fig. 2). Furthermore, responding patients had a significantly longer PFS than nonresponders (median, 8.7 [95% CI, 7.0–10.4] vs. 5.2 [95% CI, 2.7–7.7] months, P = 0.002), but difference in OS was not reached (P = 0.163).

Progression-free survival according to estrogen receptor status (a) and menstruation (b)

Overall survival according to ECOG performance status (a) and menstruation (b)
Genotype frequency and their effects on objective response, PFS, and OS
Most of the 79 tested SNPs followed the Hardy–Weinberg equilibrium (HWE), except for CYP4F11 rs1060463 G>A (P < 0.001), CYP19A1 rs700518 A>G (P = 0.018), CYP7B1 rs6987861 G>A (P = 0.015), CYP7A1 rs12542233 C>T (P = 0.029), and CYP2A6 rs28399433 T>G (P = 0.002). The observed minor allele frequency of the 79 SNPs in our study population were shown in supplementary data Table S1.
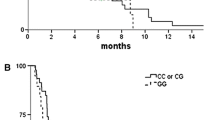
In the univariate analysis, no significant association was detected between genotype and objective response or OS after Bonferroni correction (P = 0.0006). Only one SNP, CYP1A1 rs1048943 A>G (Ile462Val), was significantly associated with PFS. Patients carrying CYP1A1 rs1048943 GA/GG genotype had a significantly longer PFS than AA genotype (median, 8.3 [95% CI, 4.3–12.0] vs. 5.3 [95% CI, 3.6–7.0] months, P = 0.0003, shown in Fig. 3). Although patients with GA/GG genotype also showed a longer OS than AA genotype (median, 20.8 [95% CI, 19.3–22.3] vs. 15.7 [95% CI, 10.7–20.7] months, P = 0.035), the threshold P value of 0.0006 was not reached.

Progression-free survival according to CYP1A1 rs1048943 A>G polymorphism
To evaluate the risk of disease progression, we carried out a Cox regression analysis entering known clinical factors (menstruation and estrogen receptor status) and polymorphism (CYP1A1 rs1048943 A>G) that significantly associated with PFS in the univariate model. In this model, we found that estrogen receptor and menstruation status were not independent predictors of PFS, while CYP1A1 rs1048943 A>G polymorphism resulted a significant predictor (P = 0.004, Table 2).
Because no SNP in our study was associated with OS, the Cox regression analysis of OS only included known clinical factors (ECOG performance status and menstruation status) that significantly associated with OS in the univariate model. In this model, we found that ECOG performance status was an independent predictor of OS (P < 0.001), while menstruation status not (P = 0.137).
Discussion
As is widely accepted, breast cancer is a heterogeneous disease with different morphologies, molecular biological behaviors, response to treatment, and prognosis (Mehta et al. 2011). The concept of “personalized treatment” recognizes these heterogeneities and emphasizes that each patient should be treated uniquely. Under this new paradigm, intensive efforts have been made to investigate appropriate biomarkers for treatment outcomes. Up to now, a lot of markers have been identified in the treatment of breast cancer: immunoglobulin G fragment C receptor polymorphisms for response to trastuzumab (Musolino et al. 2008), CYP2D6 genotype for clinical outcome of tamoxifen (Goetz et al. 2005; Schroth et al. 2007), fibroblast growth factor receptor 4 Arg388 genotype for resistance to adjuvant therapy (Thussbas et al. 2006), p53 Arg72Pro polymorphism for pathologic response to neoadjuvant chemotherapy (Xu et al. 2005), and so on.
Our study tried to explain inter-individual difference in clinical outcome from pharmacogenetic aspects. We prospectively assessed 79 genetic polymorphisms in CYP450, whose MAF were ≥10%, in 69 MBC patients who received docetaxel plus capecitabine chemotherapy. Although we are unable to find any prognostic impact of CYP450 polymorphism on objective response or OS, one SNP (CYP1A1 rs1048943 A>G) was shown to be related to PFS in the univariate analysis (P = 0.0003). In the multivariate analysis, after adjusting clinical factors associated with PFS in the univariate analysis, the association remained significant (P = 0.004). This SNP was also marginally related to OS (P = 0.035), and did not pass the threshold P value of 0.0006. It seems that it is a potential biomarker for survival outcome after docetaxel plus capecitabine chemotherapy for MBC.
CYP1A1 rs1048943 A>G substitution at codon 462 in exon 7 can result in amino change from isoleucine to valine near the heme-binding region and increases CYP1A1 activity by twofold (Cosma et al. 1993). It is possible that the elevated CYP1A1 enzyme activity leads to altered drug metabolism and difference prognosis. Future investigations should validate this result and clarify the underlying mechanisms. A lot of researches are interested in CYP1A1 rs1048943 A>G, and this SNP has been reported to be associated with the risk of lung cancer (Chen et al. 2011), breast cancer (Sergentanis and Economopoulos 2010), non-Hodgkin lymphoma (Kim et al. 2009), esophageal cancer (Zhuo et al. 2009), chronic myeloid leukemia (Taspinar et al. 2008), colorectal cancer (Kiss et al. 2007), gallbladder cancer (Tsuchiya et al. 2007), prostate cancer (Yang et al. 2006; Murata et al. 2001), and epithelial ovarian cancer (Aktas et al. 2002).
In this study, we focused on SNPs which are inherited genetic variations harbored by all the cells. They can easily be genotyped in blood and are more convenient to adopt into clinical practice than tumor expression profiles, which need biopsies of patients’ tumors. Many studies have been conducted to analyze SNPs in CYP450 in order to establish their possible relationship with chemotherapeutic outcomes in patients with breast cancer. CYP3A4 rs2740574 A>G polymorphism has been reported to be independently impact survival after adjuvant chemotherapy for node positive breast cancer (Gor et al. 2010). CYP2B6 rs8192709 C>T (Arg22Cys) and rs12721655 A>G (Lys139Glu) polymorphisms affect response to doxorubicin and cyclophosphamide therapy (Bray et al. 2010). CYP1B1 rs1056836 C>G (Leu432Val) polymorphism can influence progression-free survival of paclitaxel-based chemotherapy (Marsh et al. 2007). Our study adds more information between genetic polymorphism and chemotherapeutic outcomes. We identify CYP1A1 rs1048943 A>G (Ile462Val) as an independent predictor for progression-free survival after docetaxel plus capecitabine chemotherapy in patients with MBC.
In our study, 5 SNPs significantly deviated from the HWE. The reasons may be genotyping errors, patient selection bias, or that these SNPs are associated with the risk of breast cancer. As far as we know, there is no report whether the five SNPs are associated with the incidence of breast cancer. However, polymorphism of CYP2A6 rs28399433 T>G has been observed to be associated with altered risk of upper aerodigestive tract cancer (Canova et al. 2009). None of these five SNPs were associated with objective response, PFS, or OS.
The strength of our study included prospective analysis, detailed clinical information, focus on all SNPs with MAF ≥10% in CYP450, and conservative statistical methods for multiple comparisons. Our study also had several limitations. First, even though the present data identified CYP1A1 rs1048943 A>G (Ile462Val) as a statistically significant prognostic factor for survival outcome in MBC receiving docetaxel plus capecitabine chemotherapy, these results should be interpreted cautiously. Due to limited sample size, the result should be validated in a larger patients’ cohort to avoid any possibility of chance results. Second, in this study, we only genotyped SNPs in CYP450 whose MAF were ≥10%. Those uncommon SNPs whose MAF <10% could also play a determinant role in clinical outcome. Again, our study only included SNPs in CYP450. Investigation of polymorphisms in other genes, such as drug transporter and drug targets, might provide more information for individually optimized chemotherapy.
In conclusion, our study identifying the strong association between CYP450 gene variants and survival outcome represents a step toward personalized and optimized palliative chemotherapy in MBC. Large prospective studies are needed to validate this finding.
References
Aktas D, Guney I, Alikasifoglu M et al (2002) CYP1A1 gene polymorphism and risk of epithelial ovarian neoplasm. Gynecol Oncol 86:124–128
Bray J, Sludden J, Griffin MJ et al (2010) Influence of pharmacogenetics on response and toxicity in breast cancer patients treated with doxorubicin and cyclophosphamide. Br J Cancer 102:1003–1009
Canova C, Hashibe M, Simonato L et al (2009) Genetic associations of 115 polymorphisms with cancers of the upper aerodigestive tract across 10 European countries: the ARCAGE project. Cancer Res 69:2956–2965
Chan S, Romieu G, Huober J et al (2009) Phase III study of gemcitabine plus docetaxel compared with capecitabine plus docetaxel for anthracycline-pretreated patients with metastatic breast cancer. J Clin Oncol 27:1753–1760
Chen Z, Li Z, Niu X et al (2011) The effect of CYP1A1 polymorphisms on the risk of lung cancer: a global meta-analysis based on 71 case-control studies. Mutagenesis 26:437–446
Cosma G, Crofts F, Taioli E et al (1993) Relationship between genotype and function of the human CYP1A1 gene. J Toxicol Environ Health 40:309–316
Goetz MP, Rae JM, Suman VJ et al (2005) Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 23:9312–9318
Gor PP, Su HI, Gray RJ et al (2010) Cyclophosphamide-metabolizing enzyme polymorphisms and survival outcomes after adjuvant chemotherapy for node-positive breast cancer: a retrospective cohort study. Breast Cancer Res 12:R26
Harbeck N, Salem M, Nitz U et al (2010) Personalized treatment of early-stage breast cancer: present concepts and future directions. Cancer Treat Rev 36:584–594
Ingelman-Sundberg M (2004) Pharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and future. Trends Pharmacol Sci 25:193–200
Ingelman-Sundberg M, Sim SC, Gomez A et al (2007) Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther 116:496–526
Jemal A, Bray F, Center MM et al (2011) Global cancer statistics. CA Cancer J Clin 61:69–90
Justenhoven C, Winter S, Hamann U et al (2010) The frameshift polymorphism CYP3A43_74_delA is associated with poor differentiation of breast tumors. Cancer 116:5358–5364
Kim HN, Kim NY, Yu L et al (2009) Polymorphisms of drug-metabolizing genes and risk of non-Hodgkin lymphoma. Am J Hematol 84:821–825
Kiss I, Orsós Z, Gombos K et al (2007) Association between allelic polymorphisms of metabolizing enzymes (CYP 1A1, CYP 1A2, CYP 2E1, mEH) and occurrence of colorectal cancer in Hungary. Anticancer Res 27:2931–2937
Kiyotani K, Mushiroda T, Imamura CK et al (2010) Significant effect of polymorphisms in CYP2D6 and ABCC2 on clinical outcomes of adjuvant tamoxifen therapy for breast cancer patients. J Clin Oncol 28:1287–1293
Ma X, Qi X, Chen C et al (2010) Association between CYP19 polymorphisms and breast cancer risk: results from 10,592 cases and 11,720 controls. Breast Cancer Res Treat 122:495–501
Marsh S, Somlo G, Li X et al (2007) Pharmacogenetic analysis of paclitaxel transport and metabolism genes in breast cancer. Pharmacogenomics J 7:362–365
Mehta R, Jain RK, Badve S (2011) Personalized medicine: the road ahead. Clin Breast Cancer 11:20–26
Michalaki V, Gennatas S, Gennatas K (2009) Low-dose capecitabine plus docetaxel as first-line therapy for metastatic breast cancer: phase II results. Anticancer Drugs 20:204–207
Moreno-Aspitia A, Perez EA (2009) Anthracycline- and/or taxane-resistant breast cancer: results of a literature review to determine the clinical challenges and current treatment trends. Clin Ther 31:1619–1640
Murata M, Watanabe M, Yamanaka M et al (2001) Genetic polymorphisms in cytochrome P450 (CYP) 1A1, CYP1A2, CYP2E1, glutathione S-transferase (GST) M1 and GSTT1 and susceptibility to prostate cancer in the Japanese population. Cancer Lett 165:171–177
Musolino A, Naldi N, Bortesi B et al (2008) Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol 26:1789–1796
O’Shaughnessy J, Miles D, Vukelja S et al (2002) Superior survival with capecitabine plus docetaxel combination therapy in anthracycline-pretreated patients with advanced breast cancer: phase III trial results. J Clin Oncol 20:2812–2823
Petros WP, Hopkins PJ, Spruill S et al (2005) Associations between drug metabolism genotype, chemotherapy pharmacokinetics, and overall survival in patients with breast cancer. J Clin Oncol 23:6117–6125
Ruiter R, Bijl MJ, van Schaik RH et al (2010) CYP2C19*2 polymorphism is associated with increased survival in breast cancer patients using tamoxifen. Pharmacogenomics 11:1367–1375
Schroth W, Antoniadou L, Fritz P et al (2007) Breast cancer treatment outcome with adjuvant tamoxifen relative to patient CYP2D6 and CYP2C19 genotypes. J Clin Oncol 25:5187–5193
Sergentanis TN, Economopoulos KP (2010) Four polymorphisms in cytochrome P450 1A1 (CYP1A1) gene and breast cancer risk: a meta-analysis. Breast Cancer Res Treat 122:459–469
Taspinar M, Aydos SE, Comez O et al (2008) CYP1A1, GST gene polymorphisms and risk of chronic myeloid leukemia. Swiss Med Wkly 138:12–17
Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Thussbas C, Nahrig J, Streit S et al (2006) FGFR4 Arg388 allele is associated with resistance to adjuvant therapy in primary breast cancer. J Clin Oncol 24:3747–3755
Tsuchiya Y, Kiyohara C, Sato T et al (2007) Polymorphisms of cytochrome P450 1A1, glutathione S-transferase class mu, and tumour protein p53 genes and the risk of developing gallbladder cancer in Japanese. Clin Biochem 40:881–886
Watson RG, McLeod HL (2011) Pharmacogenomic contribution to drug response. Cancer J 17:80–88
Xu Y, Yao L, Ouyang T et al (2005) p53 Codon 72 polymorphism predicts the pathologic response to neoadjuvant chemotherapy in patients with breast cancer. Clin Cancer Res 11:7328–7333
Yang J, Qian LX, Wu HF et al (2006) Genetic polymorphisms in the cytochrome P450 1A1 and 2E1 genes, smoking, drinking and prostate cancer susceptibility: a case-control study in a Han nationality population in Southern China. Int J Urol 13:773–780
Zhuo WL, Zhang YS, Wang Y et al (2009) Association studies of CYP1A1 and GSTM1 polymorphisms with esophageal cancer risk: evidence-based meta-analyses. Arch Med Res 40:169–179
Acknowledgments
This study was supported by Chinese National Science and Technology Major Project, Mega-Project for New Drugs Development (2011ZX09302-001-02); and Medical Oncology Leadership Fund of Beijing Municipal Government Health Bureau (2009-2-16). We thank Sherry Gu, Ph.D., from Clinical Trial Center of Duke University for her assistance with the statistical analysis.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Dong, N., Yu, J., Wang, C. et al. Pharmacogenetic assessment of clinical outcome in patients with metastatic breast cancer treated with docetaxel plus capecitabine. J Cancer Res Clin Oncol 138, 1197–1203 (2012). https://doi.org/10.1007/s00432-012-1183-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-012-1183-5