
Abstract
Introduction
MiRNAs are short single stranded RNAs that are associated with gene regulation at the transcriptional and translational level. Changes in their expression were found in a variety of human cancers. In the present study, we aimed to investigate expression profiles of miRNA (miRNA) in the clear cell subtype of kidney cancer and to develop further understanding of the molecular mechanisms involved in the pathogenesis of renal cell carcinoma.
Materials and methods
We analyzed the miRNA expression profiles in 30 pairs of renal cell carcinoma and adjacent nontumorous tissue (NT), using a mammalian miRNA microarray containing whole human mature and precursor miRNA sequences. Real-time quantitative PCR was performed to confirm the array results.
Results
The miRNA microarray chip analysis identified 86 miRNAs differentially expressed in renal cell carcinoma tissues and a total of 38 miRNAs exhibited higher expression in the renal cell carcinoma samples than that in the NT samples, while 48 miRNA demonstrated lower expression in the renal cell carcinoma samples than that in the NT samples.
Conclusion
Quantitative real-time PCR analysis confirmed microarray data. The report supports that many miRNA expressions were altered in renal carcinoma, whose expression profiling may provide a useful clue for the pathophysiology research. However, further longer-term researches are required to investigate the relationship between miRNA and renal carcinoma as well as their role in carcinogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Around 208,500 new cases of kidney cancer are diagnosed in the world each year, accounting for just a little less than 2% of all cancers. Renal cell carcinoma is a kidney cancer that originates in the lining of the proximal renal tubule, the very small tubes in the kidney that filter the blood and remove waste products. Renal cell carcinoma is the most common type of kidney cancer, and the most common type in adults, responsible for approximately 80% of cases.
MiRNAs constitute a large class of small regulatory RNAs (Bartel 2004). Their mechanism of action and the scope of their biological roles are beginning to be understood. The mature miRNA binds to the 3′ untranslated region of the target mRNA through imperfect base pairing, producing translational arrest and/or degradation of the mRNA. Evidence indicates that miRNAs play a role in the pathogenesis of human disorders including cancer (Chiosea et al. 2007; Ozen et al. 2008). DNA, RNA or protein profile of carcinoma research has not successfully elucidated the pathogenesis of kidney carcinoma. Until now, only few data are available on miRNAs in renal cell carcinoma. So this study attempts to reveal a difference between the miRNA profile of clinical specimens of renal cell carcinoma and adjacent benign tissue to develop further understanding of the molecular mechanisms involved in the pathogenesis of kidney carcinoma.
Patients and methods
Human subjects
Thirty pairs of clear cell subtype of kidney cancer and adjacent benign renal tissue from patients (Table 1) were available for the study analysis. This study was performed according to the guidelines of Weifang Medical University, which abides by the Helsinki Declaration on ethical principles for medical research involving human subjects. All subjects gave informed consent to the work. For each case, tumor samples with matched adjacent benign tissue were collected during surgical resections and stored in liquid nitrogen. Sections from each specimen were examined by a pathologist and graded histologically (Fig. 1).

Histopathologic image of cell carcinoma of the nephrectomy specimen. Tissues were collected at the indicated time, fixed in l0% neutral buffered formalin, embedded in paraffin and then stained with hematoxylin and eosin (H&E) for histology examination. Data shown were a representative experiment (magnification ×200)
RNA isolation and RNA quality control
Tissues were cryopulverized using the Biopulverizer™ (Biospec, USA) and homogenized using the Mini-Bead-Beater-16. Total RNA was isolated from the tissues using TRIzol® Reagen (Invitrogen life technologies) and further purified using a RNeasy mini kit (Qiagen, Denmark) according to the manufacturer’s instructions. The concentration and quality of RNA from each patient tissue were measured by NanoDrop Specthophotometer and checked by gel electrophoresis. The equal amount RNA from each carcinoma sample and benign sample were pooled in two groups (tumor group and normal group), respectively.
MiRNA labeling and miRNA array hybridization
MiRNA expression profiles were carried out and generated from the two pooled total RNA groups above mentioned by applying the miRCURY locked nucleic acid (LNA) microarray platform (Exiqon, Denmark) (Dai et al. 2007). All procedures were carried out according to manufacturer’s protocol. Briefly, 1 μg total RNA was labeled with the Hy3™ or Hy5™ fluorophores, using miRCURY™ Array Power Labeling kit (Exiqon, Denmark). After stopping the labeling procedure, briefly spin the reaction and leave it at 4°C. The two samples from the Hy3™ and Hy5™ labeling reactions are combined on ice. The samples were hybridized on a hybridization station using miRCURY™ LNA miRNA Array (v.11.0) containing Tm –normalized probes for 847 human miRNAs. Microarrays with labeled samples were hybridized at 56°C for overnigh using a heat-shrunk hybridization bag and washed using miRCURY Array Wash buffer kit (Exiqon, Denmark). After hybridization, the chip slides were washed, dried and scanned immediately. Each miRNA spot was replicated for four times on the same slide and two microarray chips have been used for each group.
Scanning and data analysis
Scanning is performed with the Axon GenePix 4000B microarray scanner. GenePix pro V6.0 is used to read the raw intensity of the image. In order to produces the best within-slide normalization to minimize the intensity-dependent differences between the dyes, signal intensities for each spot were scanned and calculated by subtracting local background (based on the median intensity of the area surrounding each spot) from total intensities using locally weighted scatter plot smoothing (Lowess) Normalization (MIDAS, TIGR Microarray Data Analysis System). After normalization, obtained average values for each miRNA spot were used for statistics. The ratio of green signal to red signal was calculated. The threshold value we used to screen up- and down-regulated miRNAs is fold change >2.00 and fold change <0.50. Hierarchical clustering for differentially expressed miRNAs was generated using standard correlation as a measure of similarity.
Real-time quantitative PCR
Real-time quantitative PCR was performed to confirm the array results. Reverse transcriptase reactions contained 600 ng of purified total RNA, 20 nM stem-loop RT primer (Table 2), 1 × RT buffer, 0.125 mM each of dATP, dGTP, dCTP and dTTP, 1U/μl reverse transcriptase and 0.6 U/μl RNase Inhibitor. Using the Gene Amp PCR System 9700 (Applied Biosystems, USA), 20 μl reactions were performed with the following thermal cycling parameters: 30 min at 16°C, 42 min at 42°C, 5 min at 85°C and then held at 4°C. Each reaction mixture for real-time quantitative PCR contained 1 × PCR buffer, 1.5 mM MgCl2, 0.25 mM each of dATP, dGTP, dCTP and dTTP, 1 U DNA polymerase, 0.4 μM of each primer (Table 2), 0.25 × SYBR Green I, 1 μl cDNA, and deionized water to a total volume of 25 μl. Reactions were run with the following thermal cycling parameters: 95°C for 5 min followed by 35 cycles of 95°C for 10 s and 60°C for 60 s. The threshold cycle (Ct) is defined as the fractional cycle number at which the fluorescence passes the fixed threshold, and each sample was normalized on the basis of its endogenous U6 RNA content. The experiment was conducted in triplicate.
Statistical analysis
Data were presented as mean ± standard deviation (SD). ANOVA tests or Student’s t tests were used for statistical analysis. The data were regarded as significantly different at P < 0.05.
Results
The quantity and quality of the RNA
RNA from each patient tissue were checked by gel electrophoresis, absorbance at A260/280 ratio and A260/230 ratio, respectively. For spectrophotometer, the O.D. A260/A280 ratios were between 1.8 and 2.1. The O.D. A260/A230 ratios were more than 1.8. The 28S and 18S ribosomal RNA bands were fairly sharp, intense bands. A260/230 ratio and gel electrophoresis results confirmed the good quality of RNA isolation (data not shown). RNA quantification from each patient tissue was insured by NanoDrop ND-1000 instrument. The equal amount RNA from each carcinoma samples and from matched benign samples were pooled in the two group, respectively (tumor group and normal group). MiRNA expression profiles were carried out and generated from the two groups.
Differential expression of miRNAs between normal and tumor tissues
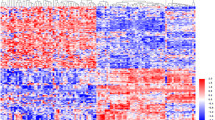
By employing a highly sensitive, high-throughput and specific miRCURY™ LNA microarray platform, miRNA expression profiles for the two groups were determined. After normalization, obtained average values for each miRNA spot were used for statistics. The ratio of red signal to green signal was calculated. The statistical significance of differentially expressed miRNA was considered to have significant differential expression if they were up- or down-regulated at least twofold. A total of 847 miRNAs were detected by miRNA microarray in the two groups. The microarray-based experiments identified 38 overexpressed and 48 down-regulated miRNAs in malignant samples. Expression in renal cell carcinoma samples compared to matched nonmalignant samples was increased on average by 2.1- to 93-fold, but decreased by 4.9- to 16-fold (Tables 3, 4). The P values for these 86 miRNAs were less than 0.05 in tumor tissue compared with normal tissue. Based on these differentially expressed miRNAs, a tree with clear distinction between tumor tissue and normal tissue was generated by cluster analysis (Fig. 2).

Hierarchical clustering of miRNA in kidney tissue samples. Kidney tissue samples were clustered according to the expression profile of 86 differentially expressed miRNAs between tumor tissue and normal tissue. Data from each miRNA were median centered. Samples are in columns and miRNAs in rows. N normal, T tumor. The P values for these miRNAs were less than 0.05 in tumor tissue compared with normal tissue
Validation of microarray results using real-time PCR
To validate our microarray results, quantitative RT–PCR analysis of normal and cancer tissue was performed for hsa-miR-16, hsa-miR-451, hsa-miR-141 and hsa-miR-200c. The RT-PCR analysis is consistent with microarray data (Fig. 3): expression of hsa-miR-16 and hsa-miR-451 was up-regulated and hsa-miR-141 and hsa-miR-200c was down-regulated in kidney cancer tissues.

Confirmation miRNA expression by quantitative real-time PCR. Quantitative real-time PCR analysis confirmed microarray data: miR-16 and miR-451 were up-regulated; miR-141 and miR-200c were down-regulated in kidney cancer tissue. The experiment was conducted in triplicate. # P < 0.05 versus normal
Genomic location of differentially expressed miRNAs
Chromosomal locations of miRNA genes have frequently provided important insight into the roles of miRNAs in specific diseases. Chromosomal distribution of some differentially expressed miRNAs in the human genome was also determined using miRBase: Sequence Release 10. 1 (Griffiths-Jones et al. 2006, 2008). By mapping miRNA genes onto the chromosomes, genes encoding differentially expressed miRNAs were found on all chromosomes but the 8, 10, 11, 21, 22 and Y chromosomes. Of all the chromosomes, chromosomes 1, 9, 14, 17 and X appear to contain the most differentially expressed miRNA genes (a total of 26 miRNAs). The most down-regulated miRNA gene, hsa-miR-141, was located on chromosome 12. The second up-regulated miRNA gene, hsa-miR-451, was located on chromosome 17.
The miRCURY™ LNA miRNA Array (v.11.0) contains 435 miRPlus™ capture probes for detection of miRNAs not included in miRBase. These probes give researches unique access to information about miRNAs otherwise not available. Our result showed that a total of 32 hsa-miRPlus were found to be differentially expressed in cancer tissue compared to normal kidney tissue, with 13 increased and 19 decreased in cancer tissue. The report supports that many hsa-miRPlus expressions were altered in renal carcinoma, however, further longer-term investigations are required to investigate the relationship between them and renal carcinoma as well as their function in carcinogenesis.
Discussion
MiRNAs constitute a recently identified class of small endogenous noncoding RNAs that act as negative regulators of the protein-coding gene expression and may impact cell differentiation, proliferation and survival, i.e., all fundamental cellular processes implicated in carcinogenesis.
Kidney cancer is any cancer that originates in kidney. Renal cell carcinoma is by far the most common type of kidney cancer. It accounts for about 9 out of 10 kidney cancers. DNA, RNA or protein profile of kidney carcinoma research has not successfully elucidated the pathogenesis of kidney carcinoma. Until now, there is few report of kidney carcinoma research from the aspect of miRNA. In the present study, we aimed to analyze the miRNA expression profiles in renal cell carcinoma and adjacent nontumorous tissue (NT) and to develop further understanding of the molecular mechanisms involved in the pathogenesis of kidney carcinoma.
In the present study, we comprehensively isolated and analyzed miRNAs in renal cell carcinoma tissue and normal kidney tissues using high-throughput miRNA microarray analysis. The comparison of miRNA levels between kidney cancer tissue and normal kidney tissue in Tables 3 and 4 gives some information about the kidney cancer and miRNAs. The location of some of these miRNAs on chromosome and their accession number in miRBase were showed in Tables 3 and 4. Some of these miRNAs have been identified as tumor suppressors through their ability to control several oncogenic pathways and play an important role in kidney carcinogenesis (Ortholan et al. 2009).
Forty-three miRNAs were reported as differentially expressed between renal cell carcinoma and normal kidney by Nakada et al. using a miRNA microarray platform which covers a total of 470 human miRNAs (Sanger miRBase release 9.1), of which 37 were significantly down-regulated in renal cell carcinoma and the other 6 were up-regulated. In the present study, we found that 86 miRNAs were differentially expressed between renal cell carcinoma and normal kidney tissue using a miRNA microarray platform which covers a total of 847 human miRNAs (Sanger miRBase release 11.0), of which 48 were significantly down-regulated in renal cell carcinoma and 38 were up-regulated. We observed that hsa-miR-141 was the most significantly down-regulated miRNAs in renal cell carcinoma and hsa-miR-200c was also down-regulated in comparison with normal kidney tissue, which is consistent with the result of Nakada et al. ZFHX1B mRNA, a transcriptional repressor for CDH1/E-cadherin, is a hypothetical target of both hsa-miR-141 and hsa-miR-200c. Overexpression of hsa-miR-141 and hsa-miR-200c caused down-regulation of ZFHX1B and up-regulation of E-cadherin in renal carcinoma cell lines and lung cancer cells (Hurteau et al. 2007; Nakada et al. 2008). These data showed that hsa-miR-141 and hsa-miR-200 c may have a tumor suppressor function in cancer cells and could be a promising treatment in anticancer therapy.
Hsa-miR-16, located at chromosome 13q14, is involved in cell proliferation and apoptosis regulation; it may interfere with either oncogenic or tumor suppressor pathways. Jung et al. (2009) found that hsa-miR-16 was increased in kidney cancer. We also found that has-miR-16 was up-regulated in renal cell carcinoma. Tsang and Kwok (2009) reported that one of has-miR-16 target genes is confirmed to be the protein Bcl-2. Transfection with anti-miR-16 inhibitor could suppress miR-16 expression, up-regulated Bcl-2 expression and induce apoptosis in cancer cells. A single miRNA can target several 100 mRNAs, leading to a complex metabolic network. Kaddar et al. (2009) demonstrated that miR-16 can also regulate the two new targets HMGA1 and caprin-1 involved in cell proliferation.
The second up-regulated miRNA gene in renal cell carcinoma tissue, hsa-miR-451, was located on chromosome 17. However, our result is contradicted with the following results. Bandres et al. (2009) reported that hsa-miR-451 was significantly underexpressed in gastric and colorectal cancer versus nontumoral tissues, has-miR-451 is a tumor suppressor and could inhibit cell proliferation. Bandres et al. (2009) identified the novel oncogene macrophage migration inhibitory factor (MIF) as a potential target of hsa-miR-451 and also found a significant inverse correlation between has-miR-451 and MIF expression in tumoral gastric biopsies. Further research is necessary for exploring the mechanism by which miR-451 shows different expressions in different tumors above mentioned.
Chromosomal locations of miRNA genes have frequently provided important insight into the roles of miRNAs in specific diseases (Lian et al. 2009). By mapping differentially expressed miRNA genes onto the chromosomes, genes encoding differentially expressed miRNAs were found on all chromosomes but the six chromosomes. Of all the chromosomes, chromosomes 1, 9, 14, 17 and X appear to contain the most differentially expressed miRNA genes.
Our findings about some hsa-miRNAs, such as hsa-miR-126, hsa-miR-424, hsa- miR 200c, and hsa-miR-145 were consistent with the result of the recently presented works (Chow et al. 2009; Huang et al. 2009; Petillo et al. 2009). However, some hsa-miRNAs have different expressions from one another in these works, especially, no hsa-miRPlus were reported in these paper other than our work.
Although this report supports that many hsa-miRPlus expressions were altered in renal carcinoma, further longer-term investigations are required to investigate the relationship between them and renal carcinoma as well as their function in carcinogenesis.
The presented differential miRNA pattern provides a solid basis for further validation including functional studies identification of a growing number of other potential oncogenic or tumor suppressor miRNAs in kidney cancer is in constant progress.
Conclusion
Taken together, we identified 86 miRNAs differentially expressed in renal cell carcinoma, whose expression profiling may provide a useful clue for the pathophysiology research of kidney cancer. Our work indicates that miRNAs are potential diagnosis biomarkers and probable factors involved in the pathogenesis of kidney cancer. Further investigation is needed to clarify the roles of identified miRNAs in the pathogenesis of kidney cancer. The study of miRNAs may lead to finding their potential for improving diagnosis, prognosis and their impact on future therapeutic strategies.
References
Bandres E, Bitarte N, Arias F, Agorreta J, Fortes P, Agirre X, Zarate R, Diaz-Gonzalez JA, Ramirez N, Sola JJ, Jimenez P, Rodriguez J, Garcia-Foncillas J (2009) miRNA-451 regulates macrophage migration inhibitory factor production and proliferation of gastrointestinal cancer cells. Clin Cancer Res 15(7):2281–2290
Bartel DP (2004) MiRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2):281–297
Chiosea S, Jelezcova E, Chandran U, Luo J, Mantha G, Sobol RW, Dacic S (2007) Overexpression of Dicer in precursor lesions of lung adenocarcinoma. Cancer Res 67(5):2345–2350
Dai Y, Huang YS, Tang M, Lv TY, Hu CX, Tan YH, Xu ZM, Yin YB (2007) Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus 16(12):939–946
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ (2006) miRBase: miRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34:D140–D144
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ (2008) miRBase: tools for miRNA genomics. Nucleic Acids Res 36:D154–D158
Hurteau GJ, Carlson JA, Spivack SD, Brock GJ (2007) Overexpression of the miRNA hsa-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res 67(17):7972–7976
Jung M, Mollenkopf HJ, Grimm C, Wagner I, Albrecht M, Waller T, Pilarsky C, Johannsen M, Stephan C, Lehrach H, Nietfeld W, Rudel T, Jung K, Kristiansen G (2009) MiRNA profiling of clear cell renal cell cancer identifies a robust signature to define renal malignancy. J Cell Mol Med (epub ahead of print)
Kaddar T, Rouault JP, Chien WW, Chebel A, Gadoux M, Salles G, Ffrench M, Magaud JP (2009) Two new miR-16 targets: Caprin-1 and HMGA1, proteins implicated in cell proliferation. Biol Cell (epub ahead of print)
Lian J, Zhang X, Tian H, Liang N, Wang Y, Liang C, Li X, Sun F (2009) Altered miRNA expression in patients with non-obstructive azoospermia. Reprod Biol Endocrinol 7:13
Nakada C, Matsuura K, Tsukamoto Y, Tanigawa M, Yoshimoto T, Narimatsu T, Nguyen LT, Hijiya N, Uchida T, Sato F, Mimata H, Seto M, Moriyama M (2008) Genome-wide miRNA expression profiling in renal cell carcinoma: significant down-regulation of miR-141 and miR-200c. J Pathol 216(4):418–427
Ortholan C, Puissegur MP, Ilie M, Barbry P, Mari B, Hofman P (2009) MiRNAs and kidney cancer: new oncogenes and tumor suppressors, new prognostic factors and potential therapeutic targets. Curr Med Chem 16(9):1047–1061
Ozen M, Creighton CJ, Ozdemir M, Ittmann M (2008) Widespread deregulation of miRNA expression in human prostate cancer. Oncogene 27(12):1788–1793
Tsang WP, Kwok TT (2009) Epigallocatechin gallate up-regulation of miR-16 and induction of apoptosis in human cancer cells. J Nutr Biochem (epub ahead of print)
Acknowledgments
This work was supported by the grant from Shandong Provincial Education Department Foundation of China (No. J07WZ29) and Dr. initiation fund from Weifang Medical University. The authors thank the patients who participated in this study. The microarray experiments were performed by KangChen Bio-tech, Shanghai, China.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Z. Yi and Y. Fu contributed equally to this work.
Rights and permissions
About this article
Cite this article
Yi, Z., Fu, Y., Zhao, S. et al. Differential expression of miRNA patterns in renal cell carcinoma and nontumorous tissues. J Cancer Res Clin Oncol 136, 855–862 (2010). https://doi.org/10.1007/s00432-009-0726-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-009-0726-x