
Abstract
The intestinal microbiota (previously referred to as “intestinal flora”) has entered the focus of research interest not only in microbiology but also in medicine. Huge progress has been made with respect to the analysis of composition and functions of the human microbiota. An “imbalance” of the microbiota, frequently also called a “dysbiosis,” has been associated with different diseases in recent years. Crohn’s disease and ulcerative colitis as two major forms of inflammatory bowel disease, irritable bowel syndrome (IBS) and some infectious intestinal diseases such as Clostridium difficile colitis feature a dysbiosis of the intestinal flora. Whereas this is somehow expected or less surprising, an imbalance of the microbiota or an enrichment of specific bacterial strains in the flora has been associated with an increasing number of other diseases such as diabetes, metabolic syndrome, non-alcoholic fatty liver disease or steatohepatitis and even psychiatric disorders such as depression or multiple sclerosis. It is important to understand the different aspects of potential contributions of the microbiota to pathophysiology of the mentioned diseases.
Conclusion: With the present manuscript, we aim to summarize the current knowledge and provide an overview of the different concepts on how bacteria contribute to health and disease in animal models and—more importantly—humans. In addition, it has to be borne in mind that we are only at the very beginning to understand the complex mechanisms of host-microbial interactions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The normal microbiota of humans consists of a few eukaryotic fungi, viruses, and some archaea that colonize the lower intestinal tract [68, 79]. By far, the most prominent component of the normal microbiota, however, is bacteria [148, 154]. They are the most numerous and obvious microbial components of the normal flora. Up to 100 trillion (1014) microorganisms [166] per human colonize the intestinal tract making about 2 kg of the body weight. They represent at least 300–1000 different species [57, 166].
Interestingly, at present, no one is able to exactly determine how many bacterial species might really be represented in an intestinal microbiota probe. This is dependent on the mathematical algorithm used for the analysis and the cutoff for similarity of the 16S RNA sequence (usually 97 % sequence identity is chosen to demarcate different “species” and define a so-called operational taxonomic unit (OTU) [117]) [124]. However, the knowledge on microbial composition has greatly increased with the use of culture-independent analysis methods. Culture-dependent methods in the past have been hampered by the fact that the majority of bacterial species cannot be cultured under aerobic conditions. Anaerobic conditions are hard/impossible to maintain. Only a short contact with oxygen may kill several species thereby leading to conditions that further decrease vitality of other species. Thus, culture methods are not suited to really give us an overview over the complete intestinal flora of a human individual.
Culture-independent methods mainly employ variations in genes that are common in all bacteria with, however, species-specific differences, such as 16S RNA [46, 161, 167, 168]. While these culture-independent methods such as restriction fragment length polymorphism (RFLP) analysis or pyro-sequencing have enabled a more in-depth study of the microbial composition in the human intestine with higher precision, little is known how the complex composition is modulated and how environmental factors such as nutrition, medication use, way of life, toxins, and other exogenic factors such as smoking might change the balance between different microbial species [26, 201]. Besides environmental influences, there are other influences on the composition of the intestinal microbiota. Among those are genetic influences. There is obviously an adaptation between the genetic structure of the host and the genetic composition of the bacteria [11, 80, 137, 153, 197]. Based on the genetic background, metabolites are synthesized by the bacteria as well as the host [195]. Those metabolites will cross-react and influence each other [195], indicating that the metabolic activity of host and commensal bacteria also needs to be in a tight balance. An extremely rare though nevertheless impressive illustration of host-microbial metabolic interaction is the so-called gut fermentation syndrome (auto-brewery syndrome), where an overgrowth with specific microbes may induce considerable and clinically relevant ethanol blood levels in the absence of any alcohol intake [36]. Another aspect that influences the bacterial composition is the age of the host, the gender, the area of living, the amount of stress perceived, the extent of regular physical exercise, the climate, and other influences that have been mentioned before [120, 213].
Bacteria select their environment
It is well known that bacteria have environmental preferences and that certain bacteria only colonize certain areas of the body [66, 131, 214]. This may be due to an interaction with surface molecules on the respective tissue cells. Table 1 gives a list of specific bacterial strains and their preferred tissue of adherence.
It was well established from disease description, which was then further analyzed by molecular methods, that for example Corynebacterium diphteriae mainly colonizes the throat epithelium [125]. This is the reason why diphtheria as a disease is mainly a throat disease. On the other hand, Neisseria gonnorhoeae mainly colonizes the urogenital epithelium and this is the reason why this is a veneric disease [204]. Contrastingly, there are bacterial species such as Vibrio cholerae, the bacterium causing cholera disease, that mainly colonize the small intestinal epithelium. Staphylococcus aureus has a preference for the nasal membranes and Staphylococcus epidermidis for the skin.
Bacteria have found their ecological niche in the human body and have selected attachment molecules where they have an advantage above other bacteria [39, 62, 112]. For some of the bacterial species, both the bacterial ligands for attachment at the host cell or tissue receptor have been identified [16, 141, 184]. This is illustrated in Table 2.
Streptococcus pyogenes uses its protein F to bind to the amino terminus of fibronectin which is largely expressed on the pharyngeal epithelium [191]. Neisseria gonorrhoeae has N-methylhenyl-alanine pili that bind to glucosamine-galactose carbohydrate residues on proteins mainly in the urethral and cervical epithelium. Escherichia coli has type 1 fimbriae that bind certain carbohydrates on either the intestinal epithelium or the urethral epithelium [4, 12, 30, 211]. Their binding sites are to some extend carbohydrate specific [4, 12, 30, 211].
Certainly, there are many more bacterial ligands from bacteria that are harder to culture. Protein expression of bacterial species that cannot be cultured obviously cannot be investigated. So far, we only have cDNA sequences from those bacteria. As there is no knowledge on protein composition of the bacterial wall of those bacteria species, the host cell receptor or tissue receptor cannot be identified. Most of those bacteria live in the intestinal lumen or are attached to the intestinal epithelial cells.
In general, the forms of bacterial colonization are either mutualistic, commensalistic, or opportunistic [178]. Mutualism means that both organisms benefit from the co-existence. Most of the intestinal bacteria therefore are not commensalistic (despite the fact that they are called commensals) but mutualistic, because both, the bacteria and the human organism, benefit from their existence [11]. In a commensalistic situation, one organism benefits and the other is neither helped nor harmed. If our intestinal bacteria would be commensalistic, this would mean that they profit but the human body has no profit. In most scenarios and situations, this is not the case: The relationship between colonizing bacteria and the human body most frequently is a mutualistic one. Opportunistic would mean that under normal conditions, the microbe does not cause disease but if conditions become conductive, it can cause disease. Opportunistic infections can be induced by Staphylococcus aureus and others that usually only become infectious when they enter the body whereas there is no problem with colonization on the skin or even in the intestine.
Why do we have bacterial colonization in the intestine?
The number of bacteria found throughout the gastrointestinal tract differs from the esophagus to the rectum (Fig. 1). Whereas the number of bacteria in the esophagus and the stomach is low with 101 to 103 bacteria per milliliter, already in the upper small intestine the number of bacteria clearly increases. Per gram of small intestinal content, the number of bacteria is 103 to 104, which is still low. However, there is a steady increase of bacterial concentration toward the lower small intestine. In the ileum and in the terminal ileum, there are 108 to 109 bacteria per gram content. Finally, in the colon, there are 1012 to 1014 bacteria per gram of feces (Fig. 1).

Numbers of bacteria per segment of the GI tract in healthy individuals
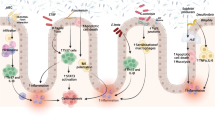
Those bacteria should not enter the body and there are several mechanisms to protect body integrity and to form a barrier against invasion of bacteria. First of all, the epithelium of the intestinal mucosa forms a monolayer with intercellular contacts that inhibit the passage of bacterial products and potential antigens through this monolayer barrier. Nevertheless, this barrier becomes leaky and single cells are extruded as shown by Alistair Watson and co-workers, causing hole-like structures in the intestinal barrier which therefore is not completely mechanically tight [121, 205, 206].
To maintain barrier integrity, several other mechanisms are necessary [60, 136, 146, 193] (Table 3). One is gut motility which prevents the long-term interaction between certain bacteria and small mucosal areas. A further important mechanism is the secretion of mucins by goblet cells as well as chloride secretion. Also, defensins are very effective in preventing the invasion of bacteria into the mucosa [207]. Defensins are like human antibiotics that can kill bacteria due to partial destruction of the cell wall. The mucin layer that is above the mechanical barrier of the epithelial cells usually contains defensins bound to the mucin structure. This mucin layer above the brush or the membrane of the epithelial cells usually is almost sterile; no bacteria are found close to the epithelial cells. However, this mucus is colonized in chronic intestinal inflammation such as Crohn’s disease or ulcerative colitis [42, 185–187] (Fig. 2). Furthermore, there is a competition between different bacterial strains at the mucosal surface. Usually, mucus-layer-attached bacteria are more host-friendly as luminal bacteria. A decreasing number of those beneficial bacteria may induce growth, adhesion, and invasion of pathogenic bacteria. This is the reason why Clostridium difficile usually only can induce colitis when the number of beneficial bacteria is diminished by antibiotic treatment.

Colonization of the mucus barrier above the intestinal epithelial cells in health subjects and patients with CD. DAPI stains DNA, EUB338 is a pan-bacterial marker. The sandwich picture shows the colonization of the CD patient’s mucus layer by living bacteria
If the potential migration and translocation of bacteria across the intestinal barrier is such a big problem for the human body, why is the intestinal lumen colonized with bacteria at all? The normal flora synthesizes and excretes vitamins in excess of their own needs and contributes to vitamin delivery to the human body [80]. Among those vitamins are vitamin K, vitamin B12, and other B vitamins (see Table 4). The normal flora also prevents the colonization by pathogens. This is facilitated by competition for attachment sites or by a competition for essential nutrients. For example, important insights have been derived from Salmonella studies that clearly show that Salmonella is not very infective in a mouse that has a colon colonized with normal commensal bacteria [15]. Only when the number of bacteria is decreased by antibiotic treatment or in germ-free animals that Salmonella can already cause disease in very low numbers [15, 71, 182].
In addition, the intestinal bacteria produce a variety of substances ranging from peroxides to highly specific other metabolic products that support epithelial growth and metabolism. Without bacterial colonization, some of the digestive enzymes would not be induced sufficiently. In various animal experiments, Jeff Gordon’s group has shown that the colonization with bacteria dramatically induces genes in the epithelial cells which are mandatory for physiological digestive process [81].
Furthermore, the normal flora stimulates the development of the adaptive immune system30 and the lymphatic tissue, for example, the Peyer’s patches in the GI tract [13, 14]. The cecum of germ-free mice is enlarged and thin walled and the lymphatic structures are underdeveloped. Functioning lymphatic structures, however, are important during intestinal infections. Furthermore, the normal microbiota stimulates the production of cross-reactive antibodies (mainly IgA) which are secreted into the gut lumen [210]. As those antibodies are cross-reactive, they also prevent from bacterial infections. Moreover, microbial products may harbor a direct immune-regulatory potential, as for instance shown with polysaccharide A (PSA) produced by Bacteroides fragilis (a ubiquitous and mutualistic organism in the gut) that may modulate and correct systemic T cell deficiencies and TH1/TH2 imbalance [126] as well as T helper cell subsets and potentially other immune cell populations. Thus, the normal flora induces a protective mechanism for preventing infections of the GI tract.
Is there a specific role of the bacterial flora in childhood?
The gut is colonized by bacteria during the first hours of life. This colonization induces gene expression and subsequent functions on the intestinal mucosa that are important for digestions and nutrition [10, 80, 85]. Intestinal angiogenesis may also be regulated by the gut microbiota [181] indicating that the microbiota has also a role in local micro- and macro-circulation.
In addition, the human intestinal flora has an important role in shaping the immune system [108]. It profoundly influences the formation of lymphatic structures and the differentiation of lymphocytes [63].
The interaction of intestinal microbiota with the immune system may further be important in the prevention of allergic and atopic diseases. In children with atopic disease, again, an “imbalance” of the intestinal flora has been described [27, 144]. Interestingly, children with the presence of eczema had an even more diverse intestinal flora as compared to control subjects [133]. Children with eczema in this analysis had an increased abundance of the Clostridium clusters IV and XIVa, which are typically abundant in adults [133]. Again, it is unclear whether these finding are only associated with the disease or in any causal relationship. On the other hand, risk genes identified for atopic diseases include a number of genes relevant for the barrier function of the skin, again indicating a role for bacteria [58]. The skin microbiome is altered at least during acute exacerbations of the disease [98]. Whereas, indeed, the majority of studies point to an association between microbiota and especially the gut microbial composition and atopic diseases, no specific harmful or protective microbes could be identified so far [145].
A direct epidemiological link between the exposure to microorganisms (which are assumed to influence mutualistic microbial composition and immune system development in the host) in early childhood and prevalence of chronic immune-related disorders is suggested by the hygiene hypothesis [9]. This inverse correlation between exposure to microbes and infectious diseases in childhood has for instance been shown with the occurrence of inflammatory bowel disease and is one of the most prominent explanations for the increasing incidence in recent years [64, 149].
Which diseases are associated with changes of the bacterial flora?
There is a growing list of non-infectious diseases that have been associated with the composition of the bacterial flora. Among them are chronic inflammatory bowel diseases, namely Crohn’s disease and ulcerative colitis, metabolic syndrome, non-alcoholic steatohepatitis, irritable bowel syndrome, atherosclerosis, rheumatoid arthritis, and colorectal carcinoma. Interestingly, shifts in intestinal microbial composition have also increasingly been described in lifestyle factors, not only in disease states, such as for instance an increased gut microbial diversity associated to heavy exercise (professional rugby players) [34]. It appears plausible that this list of environmental factors having an impact on microbial composition will grow in the next years.
Inflammatory bowel disease
The two most frequent and clinically most important forms of inflammatory bowel diseases (IBDs) are Crohn’s disease and ulcerative colitis [75, 169]. They are chronic relapsing intestinal inflammations. More than 160 genetic factors (single-nucleotide polymorphisms, SNPs, in more than 160 genes are associated with an increased odds ratio to develop the disease) contribute to an increased risk to develop both diseases [90, 109]. However, these genetic risk factors are not disease specific [109]. The individual odds ratio of several genetic risk factors is low and there is clear evidence that environmental factors must contribute to the onset of both inflammatory diseases [158, 169]. Moreover, while the genetic pool largely remained stable within the last decades, incidence and prevalence have shown an impressive increase in the majority of epidemiologic studies all over the world and even more pronounced in threshold countries [127], further indicating a prominent role of environmental factors.
IBDs are multifactorial diseases but there definitely is an important role for bacteria. “Dysbiosis” has been detected in both diseases that is most pronounced when the inflammation is active. Alterations and changes of the predominant species of the fecal bacteria in the colon of IBD patients have been demonstrated by many groups [55, 92, 119, 151, 173, 183]. While some of the alterations observed may even be conflicting between studies, there are certain distinctive changes that have been reproduced in the scientific literature. For instance, a decrease of Faecalibacterium prausnitzii and butyrate-producing Roseburia hominis is clearly associated with Crohn’s disease (CD) [19, 73, 89, 119] and has interestingly also been found in healthy relatives of CD patients [73]. Usually, the changes are summarized in a reduction of “diversity” [52, 92, 128, 139]. A more “diverse” flora usually is regarded to be beneficial; subsequently, OTUs are statistically analyzed by “diversity indices”; however, in fact, we have no clue so far what this could mean on a functional level.
In addition, there is a reduced expression and biochemical changes of mucins in the colon of Crohn’s disease and ulcerative colitis patients [6, 18, 96]. Important studies demonstrated a reduced production of defensins (antimicrobial peptides that are mainly secreted by Paneth cells) [7, 32, 97, 138, 171] and some authors such as Fellermann, Wehkamp, Stange, and colleagues have called Crohn’s disease a “defensin deficiency disease” [61, 86, 208].
Fecal microbiota transplantation, as an attempt to radically address disturbed microbial composition and diversity, is discussed controversially in these diseases. While some authors and small case reports/series reported a clear benefit, others could not find a beneficial effect and even saw adverse events in IBD patients such as fever or diarrhea [5, 41, 103].
Most of the genetic risk factors for IBD are layers in the innate immune system that is responsible for the acute defense against invading bacteria [109, 110]. Among those risk genes are pattern recognition receptors such as NOD2, TLR4, CARD8, CARD9, or NLRP3 as well as autophagy genes such as ATG16L1, IRGM, and LRRK2 which destroy bacteria when they have entered the epithelial cells in the process called autophagy. Further, risk factors for Crohn’s disease can be found in the antibacterial response as for example in the defensin system (see above). Also, elements that are responsible for the maintenance of the epithelial barrier integrity (IBD5, DLG5, PDGER4, DMBT1, and XBP1) have been identified. In addition, there are aspects that mainly orchestrate the adaptive immune system, but they may be secondary to those defects in the innate immune response.
Interestingly, a number of the components that have been identified to be genetic risk factors in IBD also were found in other diseases such as systemic lupus erythematosus (PTPN22), ankylosing spondylitis (ERAP2), psoriasis (PTPN22), asthma (IBD5), type 2 diabetes (GCKR), coeliac disease (PTPN2), type 1 diabetes (PTPN22), leprosy (NOD2), rheumatoid arthritis (PTPN22), and multiple sclerosis (PTGER4, STAT3). This may indicate a link between those diseases and the intestinal microbiota [109, 110]. For instance, the critical role of NOD2 on intestinal microbial composition was revealed in a NOD2-deficient mouse model, where microbial alterations were found already at an early weaning stage [153].
In 2010, we could show that indeed an increased amount of the bacterial wall compound LPS can be found inside the lamina propria of Crohn’s disease patients that carry the NOD2 variants in their mucosa [100]. NOD2 variants also are responsible for increased risk to suffer from severe intestinal graft versus host disease after stem cell transplantation [70, 76–78, 102, 105, 159]. This further indicates that the bacteria and the bacterial invasion are crucial in the onset of mucosal inflammation. Further, evidence comes from the finding that antibacterial therapy by antibiotics might decrease the risk for intestinal inflammation [94].
Irritable bowel syndrome
The potential role of the intestinal microbiota in the pathogenesis of IBS has come into the focus of attention only in recent years. One of the reasons for this relatively long deferment might be based on the very nature of the “functional” fundament of the definition of IBS [132], rather precluding a well definable and identifiable anatomical or physiological alteration. However, at the very latest with the large double-blind randomized-controlled trial from Pimentel and colleagues using rifaximine, a non-absorbable derivate of rifamycine, a critical role of the gut microbiota in IBS was suggested by the main finding of the study. Significantly, more patients in the verum group reached the primary end point, i.e., the proportion of patients with a relief of global IBS symptoms [147]. Several years ago, one of the first studies characterizing microbial composition in patients with IBS from Finland found significant differences in the microbial composition of IBS patients, including several distinctive alterations on the level of genera, such as for instance Coprococcus, Collinsella, and Coprobacillus [93]. Further studies confirmed variations of microbial alterations in IBS patients compared to controls [29, 35, 150, 156], also observable in childhood IBS [165] suggesting—besides a new therapeutic target—also a diagnostic potential of distinctive microbial fingerprints to accurately identify IBS and differentiate this condition from other gastrointestinal diseases [150].
Colorectal cancer
The intestinal microbiota also seems to play an important role for the development of colorectal cancer (CRC). A recent, excellent review highlighted the insights we have on the role of the intestinal microbiota in CRC so far [84]. Components of the intestinal bacterial flora are thought to generate “genotoxic stress to promote genetic and epigenetic alterations” in the intestinal epithelial cells finally leading to cancer [84, 172]. Distinctive alterations in colorectal cancer microbiota composition, such as an increase of Fusobacterium sequences, have been described [101], although in these descriptive studies of microbial composition, the issue of either primary causative event or secondary phenomenon cannot be resolved. However, animal models appear to be an important means of elucidating this question. For instance, dysbiosis induced by NOD2 deficiency in mice resulted in an increased predisposition to colitis-associated dysplasia and cancer [40].
In an animal model of colorectal cancer, Jobin and coworkers provided evidence that mono-colonization with the commensal Escherichia coli NC101 promoted invasive CRC [88]. They further identified specific genes of this bacterium that are involved in the promotion of CRC: The deletion of a so-called “genotoxic island” from the DNA of this Escherichia coli strain NC101 decreased tumor multiplicity and invasion in the mouse model [8].
Other types of cancer
The impact of the microbiome on the development of liver cancer may be either direct or indirect. An indirect pathway would primarily contribute to metabolic syndrome and NASH (see below). The translocation of bacterial products across the intestinal barrier into the portal vein blood that contribute to senescence of stellate cells could be a direct contribution [174]. However, in liver cancer, deconstructing the distinctive pathogenetic role of the microbiota remains challenging, as obesity per se increases the risk for several cancers [157], including those of the liver.
Polymorphisms in the NOD2 gene which is a pattern recognition receptor and the first identified and long-known susceptibility factor for CD have been shown to be associated with an increased risk for a number of different cancers besides CRC [118] (for a meta-analysis, see [114], for a review, [25]). Among them is gastric cancer [203], but also urothelial cancer [69] or breast cancer [83]. On the other hand, there are studies that could not confirm this association [104, 194] making these findings somewhat disputable.
The gut microbiota may help to shape the immunological anti-cancer response as some anti-cancer therapies seem to lose efficacy in germ-free animals [198].
Metabolic syndrome
Whereas it might be obvious that bacteria can contribute to intestinal inflammation such as IBD, it seems to be a little bit more surprising that the microbiota also has been associated to metabolic diseases such as diabetes [1, 3, 22, 23, 47, 50, 56, 72, 209], metabolic syndrome [31, 189, 199], or non-alcoholic steatohepatitis [74, 212]. There is a worldwide pandemia of obesity and recent data show that 69 % of the US adults above 20 years are overweight or obese (Central for Disease Control; www.cdc.gov ). In general, in those patients, an increase in Firmicutes and Actinobacteria is found and a decrease in Bacteroidetes which is paralleled in human and mice [91, 111, 192].
Interestingly, the obese phenotype has shown to be transmissible via the fecal microbiota [113, 155, 192]. A microbiota extracted from obese mice and transferred to lean mice was followed by a significant weight gain in the lean mice. Unfortunately, it does not work in the opposite direction [155, 192]. Microbiota from lean mice does not cause a weight loss in obese mice. Nevertheless, the modification of the gut microbiota is already discussed as a future treatment strategy for obesity. At present, we cannot tell whether this is just hype or whether it will be a promise for the future. Preliminary data from a smaller trial in humans, rather focusing on insulin resistance than body weight, suggested, however, only a very modest effect [200].
There are distinctive changes in the composition of the major phyla. Phylum refers to a taxonomic rank in biology below the rank kingdom including Archaebacteria and Eubacteria from the domain Bacteria. Regarding phyla, there is an increase in energy extraction in Firmicutes-rich gut microbiota [192]. However, the relationship is most likely more complex than just an increased Firmicutes-to-Bacteroidetes ratio. No correlation to abundance of major phyla in structured weight loss programs in humans has been described [164]. On the other hand, the success in weight loss is higher in individuals that are rich in Bacteroides fragilis, Lactobacilli, and Bifidobacteria.
To date, knowledge on the mechanisms and host-microbial interactions behind a weight gain is sparse. Potential proposed explanations include an influence of intestinal bacteria and archaea on the expression of genes involved in energy metabolism [81]: There is a transcriptional response of epithelial genes upon bacterial colonization. When the altered genes are more closely looked at that are increased upon bacterial colonization, they are responsible for nutrient absorption, mucosal barrier fortification, xenobiotic metabolism, angiogenesis, and intestinal maturation. In addition, bacteria may mediate hormonal changes modulating satiety and energy harvest (such as for instance via G protein receptors) [106] or induce a chronic inflammatory state [28, 65, 82].
Rheumatic diseases
There is increasing evidence that the bacterial flora also may contribute to the onset of rheumatic diseases [24]. As mentioned before, IBD and rheumatic diseases share common risk loci such as PTPN2 or PTPN22 [107]. Similar to what has been described for IBD, an interaction between genetic and environmental risk factors seems to be pathophysiologically relevant in rheumatic diseases [170]. Similar to other diseases, distinctive intestinal microbial alterations in patients with rheumatoid arthritis compared to healthy humans have been described, such as a lower abundance of Bifidobacteria and bacteria of the Bacteroides-Porphyromonas-Prevotella group, Bacteroides fragilis subgroup, and Eubacterium rectale–Clostridium coccoides group [196].
Allergies and atopic diseases
The microbiota composition of children with atopic diseases has been found to be altered as compared to controls (for details see above). The “training” of the intestinal immune system by gut bacteria appears as an important step in the development of immune reactions during childhood. As previously mentioned, there are, however, no definite microbial taxa associated with the occurrence of distinctive allergic diseases. In a randomized trial of oral supplementation of a bacterial lysate (between week 5 and end of month 7), infant colonization with clostridia was shown with an increased risk of developing atopic dermatitis in the subsequent 6 months [142]. However, currently, no specific taxonomic microbial members are consistently found to be a risk factor for allergic diseases, having older siblings and mode of delivery, both factors having clearly shown to influence intestinal microbial composition and appear to modulate atopy risk [143]. This represents an indirect proof for the importance of microbial composition in the pathogenesis of atopy.
Heart disease
A recent review highlighted the important role the intestinal microbiota might have for the development of various heart diseases [160]. Patients with inflammatory bowel diseases appear to have a higher risk—interestingly above all in women—for coronary heart disease and cerebrovascular events [177] despite a lower prevalence of “classical” risk factors, indicating additional links between the gut and the cardiovascular system. An impaired intestinal barrier function followed by bacterial translocation and presence of bacterial products in the circulation may contribute to atherosclerosis and chronic heart failure (CHF) as recent data indicate [160]. This association is further suggested by an interesting study investigating microbial composition in atherosclerotic plaques that showed a clear correlation to the microbial composition of the hosts’ oral cavity [99].
Psychiatric disorders
An alternation of the composition of the human microbiota has been found in a mouse model of depression [140] as well as in patients with depressive symptoms [51, 130]. However, it has to be kept in mind that depression is associated with a number of changes in behavior such as food consumption, diet, and physical activity that may influence the composition of the gut microbiota.
Changes of the gut flora furthermore have been found in mouse models [45] as well as patients with autism [44, 115, 202]. Also, stress perception may have a significant influence on human microbiota [135].
It is too early to interpret these findings. They may, however, open a sight on diseases that seemed to have no connection to intestinal functions.
Possibly the most robust evidence for a brain modulating capacity of the intestinal microbial composition in humans so far comes from a randomized trial of Bifidobacteria supplementation, where significant changes in task performances and activity in brain regions important for the procession of emotion and sensation were observed in correlation to shifts in intestinal microbial composition in those women receiving the verum preparation [190].
What environmental factors drive the composition of the microbiota?
Nutrition certainly is responsible for some aspects of microbiota composition [2, 43, 59, 123, 148, 162, 163, 176, 215]. Individuals that change their living conditions also change the microbial composition of the intestinal flora. For example, the change from a mixed diet to a vegetarian diet causes changes in the microbial flora [95]. Moreover, the intestinal microbiota appears to only to be associated with Kwashiorkor (a disease state of severe malnutrition)—rather, transplant experiments using mouse models suggest a direct pathogenetic and causative role of microbial composition, as the phenotype appeared to be inducible by transplantation [179]. Antibiotic treatment has also been shown to induce alterations on microbial composition [48, 49, 87]. The sustainability of this effect is not fully understood. However, as the induced disturbances have been shown to increase the risk in children to develop IBD years or even decades after intake [175, 188], it appears plausible to consider long-term (potentially lifetime) shifting. Aside from these rather intuitively plausible modulating factors, other influences on microbial composition, where the causative link appears more obscure, have been identified, such as physical activity [34] or excessive alcohol use [129].
Interestingly, the microbial composition of the gut has been shown to be different in the elderly [33, 122]. So far, it remains to be established whether these differences in composition and temporal stability are directly linked to differences of the physiology in the elderly or rather a sort of summation effect from modulating factors (including environmental) during the whole human life span.
On the other hand, there are influences that only recently have been recognized. While in utero the entire organism is sterile, initial colonization of the newborn with microorganisms does not occur after birth as often incorrectly assumed but indeed during the very process of passage through the vaginal birth canal [53], a distinctive ecosystem with its own evolutionary history toward its current microbial composition dominated by Lactobacillus and Prevotella spp. [54, 152]. Thus, delivery mode, i.e., conventional vaginal birth (VB) versus Cesarean section (C-section), has a key impact on primal intestinal bacterial colonization and thus composition of the pioneer human intestinal microbiota [20, 54, 67].
It always has been reported that there is an average weight gain after smoking cessation of 7–8 kg [134]. Usually, it has been attributed to an increased food intake as an oral substitution for the smoking. However, data from one of the largest population-based primary prevention trial on this topic, the Multiple Risk Factor Intervention Trial, revealed that successful quitters gained weight despite the fact that they consumed less calories and had a healthier diet compared to the continuous smokers or recidivists, who did not gain weight [180].
Interestingly, smoking also has detrimental effects in Crohn’s disease, whereas in ulcerative colitis, smoking seems to be protective [17, 37, 38, 116]. There is a low incidence of ulcerative colitis especially early onset forms in smokers and a more severe disease course may appear after smoking cessation. Subsequently, we were interested in studying the composition of the intestinal microbiota upon smoking cessation. Thus, we performed a study in 10 healthy smoking subjects and a control group of 10 subjects that continued smoking or were non-smokers [21]. The observational period was 9 weeks, including five study visits to collect stool samples. An intensive counseling by physicians and psychologists was performed. Food diary controlled the food intake and strict adherence to smoking cessation was verified by carbon monoxide exhalation monitoring. Interestingly, using different microbial approaches to investigate microbial composition and quantify specific strains, including 454-pyrosequencing and fluorescence in situ hybridization, we found significant shifts of microbial composition from phyla to genera in the intervention group after smoking cessation (Fig. 3). In the intervention group, there was a significant increase of Firmicutes and Actinobacteria whereas there was a decrease of Proteobacteria and Baceroidetes. In contrast, in the two control groups, there was a stable microbial composition. This increase in Firmicutes and Actinobacteria was associated with the weight gain despite a stable intake of calories in those subjects undergoing smoking cessation [21].

Intestinal microbial composition (major phyla of the gut microbiota) 1 week before (t1) as well as 4 (t2) and 8 weeks (t3) after smoking cessation in the intervention group (I) compared to the non-smoking (N) and smoking (S) control groups with the same intervals but without abrupt change in smoking habits. Percentages of major phyla are shown for the three time points. A significant increase in fractions of Firmicutes and Actinobacteria and a decrease in fractions of Proteobacteria and Bacteroidetes can be observed in the intervention group with a stable composition in the control groups. On the left, a phylogenetic tree is shown, to illustrate the earlier separation of the samples in the intervention group prior and after smoking cessation, i.e., closer to the root of the tree
Summary
We have only started to understand the importance and the impact of our microbiota for health and disease. Many diseases have been associated with an imbalance or dysbiosis of the microbial composition such as IBD, rheumatoid diseases, atopic diseases, cancer, metabolic syndrome, and even psychiatric disease. So far, these findings are descriptive. A deeper understanding of the interactions will be necessary to finally come to new therapeutics to treat those chronic diseases. A promising hint that this may indeed be possible in the future is the clinical success of FMT (a radical, though very unspecific, approach) for recurrent Clostridium difficile colitis. For obvious reasons, other diseases will require more specific approaches. It will be important to neither raise exaggerated expectations nor make unbalanced or enthusiastic promises at present. We should bear in mind that we still need to further carefully elucidate the mechanisms by which the bacterial flora contributes to health and disease and learn how this knowledge must may be translated to overcome various obstacles still present in the creation of targeted microbial treatment approaches for the multitude of human diseases associated with altered microbial composition. Otherwise, the hype about the microbiota will soon be over and we will have missed an important chance.
References
Alam C, Bittoun E, Bhagwat D, Valkonen S, Saari A, Jaakkola U, Eerola E, Huovinen P, Hanninen A (2011) Effects of a germ-free environment on gut immune regulation and diabetes progression in non-obese diabetic (NOD) mice. Diabetologia 54(6):1398–1406. doi:10.1007/s00125-011-2097-5
Albenberg LG, Wu GD (2014) Diet and the intestinal microbiome: associations, functions, and implications for health and disease. Gastroenterology 146(6):1564–1572. doi:10.1053/j.gastro.2014.01.058
Alkanani AK, Hara N, Lien E, Ir D, Kotter CV, Robertson CE, Wagner BD, Frank DN, Zipris D (2014) Induction of diabetes in the RIP-B7.1 mouse model is critically dependent on TLR3 and MyD88 pathways and is associated with alterations in the intestinal microbiome. Diabetes 63(2):619–631. doi:10.2337/db13-1007
Andersson M, Bjornham O, Svantesson M, Badahdah A, Uhlin BE, Bullitt E (2012) A structural basis for sustained bacterial adhesion: biomechanical properties of CFA/I pili. J Mol Biol 415(5):918–928. doi:10.1016/j.jmb.2011.12.006
Angelberger S, Reinisch W, Makristathis A, Lichtenberger C, Dejaco C, Papay P, Novacek G, Trauner M, Loy A, Berry D (2013) Temporal bacterial community dynamics vary among ulcerative colitis patients after fecal microbiota transplantation. Am J Gastroenterol 108(10):1620–1630. doi:10.1038/ajg.2013.257
Antoni L, Nuding S, Wehkamp J, Stange EF (2014) Intestinal barrier in inflammatory bowel disease. World J Gastroenterol : WJG 20(5):1165–1179. doi:10.3748/wjg.v20.i5.1165
Antoni L, Nuding S, Weller D, Gersemann M, Ott G, Wehkamp J, Stange EF (2013) Human colonic mucus is a reservoir for antimicrobial peptides. J Crohn’s Colitis 7(12):e652–64. doi:10.1016/j.crohns.2013.05.006
Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C (2012) Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338(6103):120–123. doi:10.1126/science.1224820
Bach J (2002) The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 347(12):911–920. doi:10.1056/NEJMra020100
Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 101(44):15718–15723. doi:10.1073/pnas.0407076101
Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI (2005) Host-bacterial mutualism in the human intestine. Science 307(5717):1915–1920. doi:10.1126/science.1104816
Barnich N, Darfeuille-Michaud A (2010) Abnormal CEACAM6 expression in Crohn disease patients favors gut colonization and inflammation by adherent-invasive E. coli. Virulence 1(4):281–282. doi:10.4161/viru.1.4.11510
Barreau F, Madre C, Meinzer U, Berrebi D, Dussaillant M, Merlin F, Eckmann L, Karin M, Sterkers G, Bonacorsi S, Lesuffleur T, Hugot JP (2010) Nod2 regulates the host response towards microflora by modulating T cell function and epithelial permeability in mouse Peyer’s patches. Gut 59(2):207–217. doi:10.1136/gut.2008.171546
Barreau F, Meinzer U, Chareyre F, Berrebi D, Niwa-Kawakita M, Dussaillant M, Foligne B, Ollendorff V, Heyman M, Bonacorsi S, Lesuffleur T, Sterkers G, Giovannini M, Hugot JP (2007) CARD15/NOD2 is required for Peyer’s patches homeostasis in mice. PLoS One 2(6):e523. doi:10.1371/journal.pone.0000523
Barthel M, Hapfelmeier S, Quintanilla-Martinez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Russmann H, Hardt WD (2003) Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun 71(5):2839–2858
Bartlett AH, Park PW (2010) Proteoglycans in host-pathogen interactions: molecular mechanisms and therapeutic implications. Exp Rev Mol Med 12:e5. doi:10.1017/S1462399409001367
Beaugerie L, Massot N, Carbonnel F, Cattan S, Gendre JP, Cosnes J (2001) Impact of cessation of smoking on the course of ulcerative colitis. Am J Gastroenterol 96(7):2113–2116. doi:10.1111/j.1572-0241.2001.03944.x
Belley A, Keller K, Gottke M, Chadee K (1999) Intestinal mucins in colonization and host defense against pathogens. AmJTrop Med Hyg 60(4 Suppl):10–15
Benjamin JL, Hedin CR, Koutsoumpas A, Ng SC, McCarthy NE, Prescott NJ, Pessoa-Lopes P, Mathew CG, Sanderson J, Hart AL, Kamm MA, Knight SC, Forbes A, Stagg AJ, Lindsay JO, Whelan K (2012) Smokers with active Crohn’s disease have a clinically relevant dysbiosis of the gastrointestinal microbiota. Inflamm Bowel Dis 18(6):1092–1100. doi:10.1002/ibd.21864
Biasucci G, Benenati B, Morelli L, Bessi E, Boehm G (2008) Cesarean delivery may affect the early biodiversity of intestinal bacteria. J Nutr 138(9):1796S–1800S
Biedermann L, Zeitz J, Mwinyi J, Sutter-Minder E, Rehman A, Ott SJ, Steurer-Stey C, Frei A, Frei P, Scharl M, Loessner MJ, Vavricka SR, Fried M, Schreiber S, Schuppler M, Rogler G (2013) Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS One 8(3):e59260. doi:10.1371/journal.pone.0059260
Boerner BP, Sarvetnick NE (2011) Type 1 diabetes: role of intestinal microbiome in humans and mice. Ann N Y Acad Sci 1243:103–118. doi:10.1111/j.1749-6632.2011.06340.x
Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, Casella G, Drew JC, Ilonen J, Knip M, Hyoty H, Veijola R, Simell T, Simell O, Neu J, Wasserfall CH, Schatz D, Atkinson MA, Triplett EW (2011) Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One 6(10):e25792. doi:10.1371/journal.pone.0025792
Brusca SB, Abramson SB, Scher JU (2014) Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr Opin Rheumatol 26(1):101–107. doi:10.1097/BOR.0000000000000008
Bultman SJ (2014) Emerging roles of the microbiome in cancer. Carcinogenesis 35(2):249–255. doi:10.1093/carcin/bgt392
Candela M, Biagi E, Maccaferri S, Turroni S, Brigidi P (2012) Intestinal microbiota is a plastic factor responding to environmental changes. Trends Microbiol 20(8):385–391. doi:10.1016/j.tim.2012.05.003
Candela M, Rampelli S, Turroni S, Severgnini M, Consolandi C, de Bellis G, Masetti R, Ricci G, Pession A, Brigidi P (2012) Unbalance of intestinal microbiota in atopic children. BMC Microbiol 12:95. doi:10.1186/1471-2180-12-95
Cani PD, Possemiers S, van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck A, Lambert DM, Muccioli GG, Delzenne NM (2009) Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58(8):1091–1103. doi:10.1136/gut.2008.165886
Carroll IM, Chang YH, Park J, Sartor RB, Ringel Y (2010) Luminal and mucosal-associated intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Gut Pathog 2(1):19. doi:10.1186/1757-4749-2-19
Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CH, Stanners CP, Darfeuille-Michaud A (2009) Crohn’s disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med 206(10):2179–2189. doi:10.1084/jem.20090741
Chassaing B, Gewirtz AT (2014) Gut microbiota, low-grade inflammation, and metabolic syndrome. Toxicol Pathol 42(1):49–53. doi:10.1177/0192623313508481
Chu H, Pazgier M, Jung G, Nuccio SP, Castillo PA, de Jong MF, Winter MG, Winter SE, Wehkamp J, Shen B, Salzman NH, Underwood MA, Tsolis RM, Young GM, Lu W, Lehrer RI, Baumler AJ, Bevins CL (2012) Human alpha-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets. Science 337(6093):477–481. doi:10.1126/science.1218831
Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, Stanton C, van Sinderen D, O’Connor M, Harnedy N, O’Connor K, Henry C, O’Mahony D, Fitzgerald AP, Shanahan F, Twomey C, Hill C, Ross RP, O’Toole PW (2011) Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A 108(Suppl 1):4586–4591. doi:10.1073/pnas.1000097107
Clarke SF, Murphy EF, O’Sullivan O, Lucey AJ, Humphreys M, Hogan A, Hayes P, O’Reilly M, Jeffery IB, Wood-Martin R, Kerins DM, Quigley E, Ross RP, O’Toole PW, Molloy MG, Falvey E, Shanahan F, Cotter PD (2014) Exercise and associated dietary extremes impact on gut microbial diversity. Gut. doi:10.1136/gutjnl-2013-306541
Codling C, O’Mahony L, Shanahan F, Quigley EM, Marchesi JR (2010) A molecular analysis of fecal and mucosal bacterial communities in irritable bowel syndrome. Dig Dis Sci 55(2):392–397. doi:10.1007/s10620-009-0934-x
Cordell B, McCarthy J (2013) A case study of gut fermentation syndrome (auto-brewery) with Saccharomyces cerevisiae as the causative organism. Int J Clin Med 4(7):309–312
Cosnes J (2010) Smoking, physical activity, nutrition and lifestyle: environmental factors and their impact on IBD. Dig Dis 28(3):411–417. doi:10.1159/000320395
Cosnes J, Beaugerie L, Carbonnel F, Gendre JP (2001) Smoking cessation and the course of Crohn’s disease: an intervention study. Gastroenterology 120(5):1093–1099. doi:10.1053/gast.2001.23231
Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, Relman DA (2012) The application of ecological theory toward an understanding of the human microbiome. Science 336(6086):1255–1262. doi:10.1126/science.1224203
Couturier-Maillard A, Secher T, Rehman A, Normand S, de Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A, Gaillot O, Schreiber S, Lemoine Y, Ryffel B, Hot D, Nunez G, Chen G, Rosenstiel P, Chamaillard M (2013) NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest 123(2):700–711. doi:10.1172/JCI62236
Damman CJ, Miller SI, Surawicz CM, Zisman TL (2012) The microbiome and inflammatory bowel disease: is there a therapeutic role for fecal microbiota transplantation? Am J Gastroenterol 107(10):1452–1459. doi:10.1038/ajg.2012.93
Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF (2004) High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127(2):412–421
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ (2014) Diet rapidly and reproducibly alters the human gut microbiome. Nature 505(7484):559–563. doi:10.1038/nature12820
de Angelis M, Piccolo M, Vannini L, Siragusa S, de Giacomo A, Serrazzanetti DI, Cristofori F, Guerzoni ME, Gobbetti M, Francavilla R (2013) Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS One 8(10):e76993. doi:10.1371/journal.pone.0076993
de Theije CG, Wopereis H, Ramadan M, van Eijndthoven T, Lambert J, Knol J, Garssen J, Kraneveld AD, Oozeer R (2014) Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain Behav Immun 37:197–206. doi:10.1016/j.bbi.2013.12.005
Dekio I, Hayashi H, Sakamoto M, Kitahara M, Nishikawa T, Suematsu M, Benno Y (2005) Detection of potentially novel bacterial components of the human skin microbiota using culture-independent molecular profiling. J Med Microbiol 54(Pt 12):1231–1238. doi:10.1099/jmm. 0.46075-0
Dessein R, Peyrin-Biroulet L, Chamaillard M (2009) Intestinal microbiota gives a nod to the hygiene hypothesis in type 1 diabetes. Gastroenterology 137(1):381–383. doi:10.1053/j.gastro.2009.05.026
Dethlefsen L, Huse S, Sogin ML, Relman DA (2008) The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6(11):e280. doi:10.1371/journal.pbio.0060280
Dethlefsen L, Relman DA (2011) Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci 108(Supplement 1):4554–4561. doi:10.1073/pnas.1000087107
Devaraj S, Hemarajata P, Versalovic J (2013) The human gut microbiome and body metabolism: implications for obesity and diabetes. Clin Chem 59(4):617–628. doi:10.1373/clinchem.2012.187617
Dinan TG, Cryan JF (2013) Melancholic microbes: a link between gut microbiota and depression? Neurogastroenterol Motil : Off J Eur Gastrointest Motil Soc 25(9):713–719. doi:10.1111/nmo.12198
Docktor MJ, Paster BJ, Abramowicz S, Ingram J, Wang YE, Correll M, Jiang H, Cotton SL, Kokaras AS, Bousvaros A (2012) Alterations in diversity of the oral microbiome in pediatric inflammatory bowel disease. Inflamm Bowel Dis 18(5):935–942. doi:10.1002/ibd.21874
Dominguez-Bello MG, Blaser MJ, Ley RE, Knight R (2011) Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology 140(6):1713–1719
Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R (2010) Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci 107(26):11971–11975
Duboc H, Rajca S, Rainteau D, Benarous D, Maubert MA, Quervain E, Thomas G, Barbu V, Humbert L, Despras G, Bridonneau C, Dumetz F, Grill JP, Masliah J, Beaugerie L, Cosnes J, Chazouilleres O, Poupon R, Wolf C, Mallet JM, Langella P, Trugnan G, Sokol H, Seksik P (2013) Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62(4):531–539. doi:10.1136/gutjnl-2012-302578
Dunne JL, Triplett EW, Gevers D, Xavier R, Insel R, Danska J, Atkinson MA (2014) The intestinal microbiome in type 1 diabetes. Clin Exp Immunol 177(1):30–37. doi:10.1111/cei.12321
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA (2005) Diversity of the human intestinal microbial flora. Science 308(5728):1635–1638. doi:10.1126/science.1110591
Ellinghaus D, Baurecht H, Esparza-Gordillo J, Rodriguez E, Matanovic A, Marenholz I, Hubner N, Schaarschmidt H, Novak N, Michel S, Maintz L, Werfel T, Meyer-Hoffert U, Hotze M, Prokisch H, Heim K, Herder C, Hirota T, Tamari M, Kubo M, Takahashi A, Nakamura Y, Tsoi LC, Stuart P, Elder JT, Sun L, Zuo X, Yang S, Zhang X, Hoffmann P, Nothen MM, Folster-Holst R, Winkelmann J, Illig T, Boehm BO, Duerr RH, Buning C, Brand S, Glas J, McAleer MA, Fahy CM, Kabesch M, Brown S, McLean WH, Irvine AD, Schreiber S, Lee YA, Franke A, Weidinger S (2013) High-density genotyping study identifies four new susceptibility loci for atopic dermatitis. Nat Genet 45(7):808–812. doi:10.1038/ng.2642
Faith JJ, McNulty NP, Rey FE, Gordon JI (2011) Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 333(6038):101–104. doi:10.1126/science.1206025
Fasano A, Shea-Donohue T (2005) Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat Clin Prac Gastroenterol Hepatol 2(9):416–422. doi:10.1038/ncpgasthep0259
Fellermann K, Wehkamp J, Herrlinger KR, Stange EF (2003) Crohn’s disease: a defensin deficiency syndrome? Eur J Gastroenterol Hepatol 15(6):627–634. doi:10.1097/01.meg.0000059151.68845.88
Foster JA, Krone SM, Forney LJ (2008) Application of ecological network theory to the human microbiome. Interdiscip Perspect Infect Dis 2008:839501. doi:10.1155/2008/839501
Gaboriau-Routhiau V, Lecuyer E, Cerf-Bensussan N (2011) Role of microbiota in postnatal maturation of intestinal T-cell responses. Curr Opin Gastroenterol 27(6):502–508. doi:10.1097/MOG.0b013e32834bb82b
Gent AE, Hellier MD, Grace RH, Swarbrick ET, Coggon D (1994) Inflammatory bowel disease and domestic hygiene in infancy. Lancet 343(8900):766–767
Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E (2009) Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res 50(1):90–97. doi:10.1194/jlr. M800156-JLR200
Groisman EA, Casadesus J (2005) The origin and evolution of human pathogens. Mol Microbiol 56(1):1–7. doi:10.1111/j.1365-2958.2005.04564.x
Grönlund MM, Lehtonen OP, Eerola E, Kero P (1999) Fecal microflora in healthy infants born by different methods of delivery: permanent changes in intestinal flora after cesarean delivery. J Pediatr Gastroenterol Nutr 28(1):19–25
Guarino A, Wudy A, Basile F, Ruberto E, Buccigrossi V (2012) Composition and roles of intestinal microbiota in children. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstetricians 25 Suppl 1:63–66. doi:10.3109/14767058.2012.663231
Guirado M, Gil H, Saenz-Lopez P, Reinboth J, Garrido F, Cozar JM, Ruiz-Cabello F, Carretero R (2012) Association between C13ORF31, NOD2, RIPK2 and TLR10 polymorphisms and urothelial bladder cancer. Hum Immunol 73(6):668–672. doi:10.1016/j.humimm.2012.03.006
Gutierrez A, Scharl M, Sempere L, Holler E, Zapater P, Almenta I, Gonzalez-Navajas JM, Such J, Wiest R, Rogler G, Frances R (2014) Genetic susceptibility to increased bacterial translocation influences the response to biological therapy in patients with Crohn’s disease. Gut 63(2):272–280. doi:10.1136/gutjnl-2012-303557
Hapfelmeier S, Hardt WD (2005) A mouse model for S. typhimurium-induced enterocolitis. Trends Microbiol 13(10):497–503. doi:10.1016/j.tim.2005.08.008
Hara N, Alkanani AK, Ir D, Robertson CE, Wagner BD, Frank DN, Zipris D (2013) The role of the intestinal microbiota in type 1 diabetes. Clin Immunol 146(2):112–119. doi:10.1016/j.clim.2012.12.001
Hedin CR, McCarthy NE, Louis P, Farquharson FM, McCartney S, Taylor K, Prescott NJ, Murrells T, Stagg AJ, Whelan K, Lindsay JO (2014) Altered intestinal microbiota and blood T cell phenotype are shared by patients with Crohn’s disease and their unaffected siblings. Gut. doi:10.1136/gutjnl-2013-306226
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA (2012) Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482(7384):179–185. doi:10.1038/nature10809
Herfarth H, Rogler G (2005) Inflammatory bowel disease. Endoscopy 37(1):42–47. doi:10.1055/s-2004-826083
Holler E, Landfried K, Meier J, Hausmann M, Rogler G (2010) The role of bacteria and pattern recognition receptors in GVHD. Int J Inflamm 2010:814326. doi:10.4061/2010/814326
Holler E, Rogler G, Brenmoehl J, Hahn J, Herfarth H, Greinix H, Dickinson AM, Socie G, Wolff D, Fischer G, Jackson G, Rocha V, Steiner B, Eissner G, Marienhagen J, Schoelmerich J, Andreesen R (2006) Prognostic significance of NOD2/CARD15 variants in HLA-identical sibling hematopoietic stem cell transplantation: effect on long-term outcome is confirmed in 2 independent cohorts and may be modulated by the type of gastrointestinal decontamination. Blood 107(10):4189–4193. doi:10.1182/blood-2005-09-3741
Holler E, Rogler G, Herfarth H, Brenmoehl J, Wild PJ, Hahn J, Eissner G, Scholmerich J, Andreesen R (2004) Both donor and recipient NOD2/CARD15 mutations associate with transplant-related mortality and GvHD following allogeneic stem cell transplantation. Blood 104(3):889–894. doi:10.1182/blood-2003-10-3543
Hollister EB, Gao C, Versalovic J (2014) Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology 146(6):1449–1458. doi:10.1053/j.gastro.2014.01.052
Hooper LV, Gordon JI (2001) Commensal host-bacterial relationships in the gut. Science 292(5519):1115–1118
Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI (2001) Molecular analysis of commensal host-microbial relationships in the intestine. Science 291(5505):881–884. doi:10.1126/science.291.5505.881
Hotamisligil GS, Erbay E (2008) Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol 8(12):923–934. doi:10.1038/nri2449
Huzarski T, Lener M, Domagala W, Gronwald J, Byrski T, Kurzawski G, Suchy J, Chosia M, Woyton J, Ucinski M, Narod SA, Lubinski J (2005) The 3020insC allele of NOD2 predisposes to early-onset breast cancer. Breast Cancer Res Treat 89(1):91–93. doi:10.1007/s10549-004-1250-y
Irrazabal T, Belcheva A, Girardin SE, Martin A, Philpott DJ (2014) The multifaceted role of the intestinal microbiota in colon cancer. Mol Cell 54(2):309–320. doi:10.1016/j.molcel.2014.03.039
Ismail AS, Hooper LV (2005) Epithelial cells and their neighbors. IV. Bacterial contributions to intestinal epithelial barrier integrity. Am J Physiol Gastrointest Liver Physiol 289(5):G779–84. doi:10.1152/ajpgi.00203.2005
Jager S, Stange EF, Wehkamp J (2013) Inflammatory bowel disease: an impaired barrier disease. Langenbeck’s Arch Surg / Deutsche Gesellschaft fur Chirurgie 398(1):1–12. doi:10.1007/s00423-012-1030-9
Jernberg C, Lofmark S, Edlund C, Jansson JK (2010) Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology 156(11):3216–3223
Jobin C (2013) Microbial dysbiosis, a new risk factor in colorectal cancer? Med Sci : M/S 29(6–7):582–585. doi:10.1051/medsci/2013296010
Joossens M, Huys G, Cnockaert M, de Preter V, Verbeke K, Rutgeerts P, Vandamme P, Vermeire S (2011) Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 60(5):631–637. doi:10.1136/gut.2010.223263
Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, D’Amato M, Jong D de, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, Vos M de, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H, International, I. B. D. Genetics Consortium, Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC, Cho JH (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491(7422):119–124. doi:10.1038/nature11582
Kallus SJ, Brandt LJ (2012) The intestinal microbiota and obesity. J Clin Gastroenterol 46(1):16–24. doi:10.1097/MCG.0b013e31823711fd
Kang S, Denman SE, Morrison M, Yu Z, Dore J, Leclerc M, McSweeney CS (2010) Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis 16(12):2034–2042. doi:10.1002/ibd.21319
Kassinen A, Krogius-Kurikka L, Makivuokko H, Rinttila T, Paulin L, Corander J, Malinen E, Apajalahti J, Palva A (2007) The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects. Gastroenterology 133(1):24–33. doi:10.1053/j.gastro.2007.04.005
Khan KJ, Ullman TA, Ford AC, Abreu MT, Abadir A, Abadir A, Marshall JK, Talley NJ, Moayyedi P (2011) Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis. Am J Gastroenterol 106(4):661–673. doi:10.1038/ajg.2011.72
Kim MS, Hwang SS, Park EJ, Bae JW (2013) Strict vegetarian diet improves the risk factors associated with metabolic diseases by modulating gut microbiota and reducing intestinal inflammation. Environ Microbiol Rep 5(5):765–775. doi:10.1111/1758-2229.12079
Kindon H, Pothoulakis C, Thim L, Lynch-Devaney K, Podolsky DK (1995) Trefoil peptide protection of intestinal epithelial barrier function: cooperative interaction with mucin glycoprotein. Gastroenterology 109(2):516–523
Klag T, Stange EF, Wehkamp J (2013) Defective antibacterial barrier in inflammatory bowel disease. Dig Dis 31(3–4):310–316. doi:10.1159/000354858
Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Nisc Comparative Sequence Program, Murray PR, Turner ML, Segre JA (2012) Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome research 22(5):850–859. doi:10.1101/gr.131029.111
Koren O, Spor A, Felin J, Fåk F, Stombaugh J, Tremaroli V, Behre CJ, Knight R, Fagerberg B, Ley RE, Bäckhed F (2011) Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A 108(Suppl 1):4592–4598. doi:10.1073/pnas.1011383107
Kosovac K, Brenmoehl J, Holler E, Falk W, Schoelmerich J, Hausmann M, Rogler G (2010) Association of the NOD2 genotype with bacterial translocation via altered cell-cell contacts in Crohn’s disease patients. Inflamm Bowel Dis 16(8):1311–1321. doi:10.1002/ibd.21223
Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, Baselga J, Liu C, Shivdasani RA, Ogino S, Birren BW, Huttenhower C, Garrett WS, Meyerson M (2011) Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. doi:10.1101/gr.126573.111
Kreyenberg H, Jarisch A, Bayer C, Schuster B, Willasch A, Strahm B, Kremens B, Gruhn B, Schrauder A, Burdach S, Fuhrer M, Rossig C, Kabisch H, Schlegel PG, Stachel D, Beck JF, Mauz-Koerholz C, Chung TL, Holler E, Klingebiel T, Bader P (2011) NOD2/CARD15 gene polymorphisms affect outcome in pediatric allogeneic stem cell transplantation. Blood 118(4):1181–1184. doi:10.1182/blood-2011-05-356451
Kump PK, Grochenig HP, Lackner S, Trajanoski S, Reicht G, Hoffmann KM, Deutschmann A, Wenzl HH, Petritsch W, Krejs GJ, Gorkiewicz G, Hogenauer C (2013) Alteration of intestinal dysbiosis by fecal microbiota transplantation does not induce remission in patients with chronic active ulcerative colitis. Inflamm Bowel Dis 19(10):2155–2165. doi:10.1097/MIB.0b013e31829ea325
Lakatos PL, Hitre E, Szalay F, Zinober K, Fuszek P, Lakatos L, Fischer S, Osztovits J, Gemela O, Veres G, Papp J, Ferenci P (2007) Common NOD2/CARD15 variants are not associated with susceptibility or the clinicopathologic characteristics of sporadic colorectal cancer in Hungarian patients. BMC Cancer 7:54. doi:10.1186/1471-2407-7-54
Landfried K, Bataille F, Rogler G, Brenmoehl J, Kosovac K, Wolff D, Hilgendorf I, Hahn J, Edinger M, Hoffmann P, Obermeier F, Schoelmerich J, Andreesen R, Holler E (2010) Recipient NOD2/CARD15 status affects cellular infiltrates in human intestinal graft-versus-host disease. Clin Exp Immunol 159(1):87–92. doi:10.1111/j.1365-2249.2009.04049.x
le Poul E, Loison C, Struyf S, Springael J, Lannoy V, Decobecq M, Brezillon S, Dupriez V, Vassart G, van Damme J, Parmentier M, Detheux M (2003) Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem 278(28):25481–25489. doi:10.1074/jbc.M301403200
Lee YH, Bae SC, Choi SJ, Ji JD, Song GG (2012) The association between the PTPN22 C1858T polymorphism and rheumatoid arthritis. A meta-analysis update. Mol Biol Rep 39(4):3453–3460. doi:10.1007/s11033-011-1117-3
Lee YK, Mazmanian SK (2010) Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 330(6012):1768–1773. doi:10.1126/science.1195568
Lees CW, Barrett JC, Parkes M, Satsangi J (2011) New IBD genetics: common pathways with other diseases. Gut 60(12):1739–1753. doi:10.1136/gut.2009.199679
Lees CW, Satsangi J (2009) Genetics of inflammatory bowel disease: implications for disease pathogenesis and natural history. Exp Rev Gastroenterol Hepatol 3(5):513–534. doi:10.1586/egh.09.45
Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI (2005) Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 102(31):11070–11075. doi:10.1073/pnas.0504978102
Ley RE, Peterson DA, Gordon JI (2006) Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124(4):837–848. doi:10.1016/j.cell.2006.02.017
Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444(7122):1022–1023. doi:10.1038/4441022a
Liu J, He C, Xu Q, Xing C, Yuan Y (2014) NOD2 polymorphisms associated with cancer risk: a meta-analysis. PLoS One 9(2):e89340. doi:10.1371/journal.pone.0089340
Louis P (2012) Does the human gut microbiota contribute to the etiology of autism spectrum disorders? Dig Dis Sci 57(8):1987–1989. doi:10.1007/s10620-012-2286-1
Louis E, Michel V, Hugot JP, Reenaers C, Fontaine F, Delforge M, el Yafi F, Colombel JF, Belaiche J (2003) Early development of stricturing or penetrating pattern in Crohn’s disease is influenced by disease location, number of flares, and smoking but not by NOD2/CARD15 genotype. Gut 52(4):552–557
Lozupone CA, Knight R (2008) Species divergence and the measurement of microbial diversity. FEMS Microbiol Rev 32(4):557–578. doi:10.1111/j.1574-6976.2008.00111.x
Lubinski J, Huzarski T, Kurzawski G, Suchy J, Masojc B, Mierzejewski M, Lener M, Domagala W, Chosia M, Teodorczyk U, Medrek K, Debniak T, Zlowocka E, Gronwald J, Byrski T, Grabowska E, Nej K, Szymanska A, Szymanska J, Matyjasik J, Cybulski C, Jakubowska A, Gorski B, Narod SA (2005) The 3020insC Allele of NOD2 predisposes to cancers of multiple organs. Hereditary Cancer Clin Pract 3(2):59–63. doi:10.1186/1897-4287-3-2-59
Machiels K, Joossens M, Sabino J, de Preter V, Arijs I, Eeckhaut V, Ballet V, Claes K, van Immerseel F, Verbeke K, Ferrante M, Verhaegen J, Rutgeerts P, Vermeire S (2013) A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. doi:10.1136/gutjnl-2013-304833
Makivuokko H, Tiihonen K, Tynkkynen S, Paulin L, Rautonen N (2010) The effect of age and non-steroidal anti-inflammatory drugs on human intestinal microbiota composition. Br J Nutr 103(2):227–234. doi:10.1017/S0007114509991553
Marchiando AM, Shen L, Graham WV, Edelblum KL, Duckworth CA, Guan Y, Montrose MH, Turner JR, Watson AJ (2011) The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology 140(4):1208–1218. doi:10.1053/j.gastro.2011.01.004, e1-2
Mariat D, Firmesse O, Levenez F, Guimaraes V, Sokol H, Dore J, Corthier G, Furet JP (2009) The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol 9:123. doi:10.1186/1471-2180-9-123
Martinez-Medina M, Denizot J, Dreux N, Robin F, Billard E, Bonnet R, Darfeuille-Michaud A, Barnich N (2014) Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 63(1):116–124. doi:10.1136/gutjnl-2012-304119
Matias Rodrigues JF, von Mering C (2014) HPC-CLUST: distributed hierarchical clustering for large sets of nucleotide sequences. Bioinformatics 30(2):287–288. doi:10.1093/bioinformatics/btt657
Mattos-Guaraldi AL, Duarte Formiga LC, Pereira GA (2000) Cell surface components and adhesion in Corynebacterium diphtheriae. Microbes Infect / Institut Pasteur 2(12):1507–1512
Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL (2005) An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122(1):107–118. doi:10.1016/j.cell.2005.05.007
Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, Kaplan GG (2012) Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142(1):46–54. doi:10.1053/j.gastro.2011.10.001, e42; quiz e30
Mondot S, de Wouters T, Dore J, Lepage P (2013) The human gut microbiome and its dysfunctions. Dig Dis 31(3–4):278–285. doi:10.1159/000354678
Mutlu E, Keshavarzian A, Engen P, Forsyth CB, Sikaroodi M, Gillevet P (2009) Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol Clin Exp Res 33(10):1836–1846. doi:10.1111/j.1530-0277.2009.01022.x
Naseribafrouei A, Hestad K, Avershina E, Sekelja M, Linlokken A, Wilson R, Rudi K (2014) Correlation between the human fecal microbiota and depression. Neurogastroenterol Motil : Off J Eur Gastrointest Motil Soc. doi:10.1111/nmo.12378
Naumann M, Rudel T, Meyer TF (1999) Host cell interactions and signalling with Neisseria gonorrhoeae. Curr Opin Microbiol 2(1):62–70
Nielsen S, Nielsen DS, Lauritzen L, Jakobsen M, Michaelsen KF (2007) Impact of diet on the intestinal microbiota in 10-month-old infants. J Pediatr Gastroenterol Nutr 44(5):613–618. doi:10.1097/MPG.0b013e3180406a11
Nylund L, Satokari R, Nikkila J, Rajilic-Stojanovic M, Kalliomaki M, Isolauri E, Salminen S, de Vos WM (2013) Microarray analysis reveals marked intestinal microbiota aberrancy in infants having eczema compared to healthy children in at-risk for atopic disease. BMC Microbiol 13:12. doi:10.1186/1471-2180-13-12
O’Hara P, Connett JE, Lee WW, Nides M, Murray R, Wise R (1998) Early and late weight gain following smoking cessation in the Lung Health Study. Am J Epidemiol 148(9):821–830
O’Mahony SM, Marchesi JR, Scully P, Codling C, Ceolho AM, Quigley EM, Cryan JF, Dinan TG (2009) Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biol Psychiatry 65(3):263–267. doi:10.1016/j.biopsych.2008.06.026
Ohland CL, Macnaughton WK (2010) Probiotic bacteria and intestinal epithelial barrier function. American journal of physiology. Gastrointes Liver Physiol 298(6):G807–19. doi:10.1152/ajpgi.00243.2009
Olivares M, Laparra JM, Sanz Y (2013) Host genotype, intestinal microbiota and inflammatory disorders. Br J Nutr 109(Suppl 2):S76–80. doi:10.1017/S0007114512005521
Ostaff MJ, Stange EF, Wehkamp J (2013) Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol Med 5(10):1465–1483. doi:10.1002/emmm.201201773
Packey CD, Sartor RB (2009) Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr Opin Infect Dis 22(3):292–301. doi:10.1097/QCO.0b013e32832a8a5d
Park AJ, Collins J, Blennerhassett PA, Ghia JE, Verdu EF, Bercik P, Collins SM (2013) Altered colonic function and microbiota profile in a mouse model of chronic depression. Neurogastroenterol Motil : Off J Eur Gastrointest Motil Soc 25(9):733–e575. doi:10.1111/nmo.12153
Patti JM, Hook M (1994) Microbial adhesins recognizing extracellular matrix macromolecules. Curr Opin Cell Biol 6(5):752–758
Penders J, Gerhold K, Stobberingh EE, Thijs C, Zimmermann K, Lau S, Hamelmann E (2013) Establishment of the intestinal microbiota and its role for atopic dermatitis in early childhood. J Allergy Clinical Immunol 132(3):601–607. doi:10.1016/j.jaci.2013.05.043, e8
Penders J, Gerhold K, Thijs C, Zimmermann K, Wahn U, Lau S, Hamelmann E (2014) New insights into the hygiene hypothesis in allergic diseases: mediation of sibling and birth mode effects by the gut microbiota. Gut Microbes 5(2):239–244. doi:10.4161/gmic.27905
Penders J, Stobberingh EE, Thijs C, Adams H, Vink C, van Ree R, van den Brandt PA (2006) Molecular fingerprinting of the intestinal microbiota of infants in whom atopic eczema was or was not developing. Clin Exp Allergy : J Br Soc Allergy Clin Immunol 36(12):1602–1608. doi:10.1111/j.1365-2222.2006.02599.x
Penders J, Stobberingh EE, van den Brandt PA, Thijs C (2007) The role of the intestinal microbiota in the development of atopic disorders. Allergy 62(11):1223–1236. doi:10.1111/j.1398-9995.2007.01462.x
Peterson LW, Artis D (2014) Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14(3):141–153. doi:10.1038/nri3608
Pimentel M, Lembo A, Chey WD, Zakko S, Ringel Y, Yu J, Mareya SM, Shaw AL, Bortey E, Forbes WP (2011) Rifaximin therapy for patients with irritable bowel syndrome without constipation. N Engl J Med 364(1):22–32
Power SE, O’Toole PW, Stanton C, Ross RP, Fitzgerald GF (2014) Intestinal microbiota, diet and health. Br J Nutr 111(3):387–402. doi:10.1017/S0007114513002560
Radon K, Windstetter D, Poluda AL, Mueller B, von Mutius E, Koletzko S (2007) Contact with farm animals in early life and juvenile inflammatory bowel disease: a case–control study. Pediatrics 120(2):354–361. doi:10.1542/peds. 2006-3624
Rajilic-Stojanovic M, Biagi E, Heilig HG, Kajander K, Kekkonen RA, Tims S, de Vos WM (2011) Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome. Gastroenterology 141(5):1792–1801. doi:10.1053/j.gastro.2011.07.043
Rajilic-Stojanovic M, Shanahan F, Guarner F, de Vos WM (2013) Phylogenetic analysis of dysbiosis in ulcerative colitis during remission. Inflamm Bowel Dis 19(3):481–488. doi:10.1097/MIB.0b013e31827fec6d
Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, Brotman RM, Davis CC, Ault K, Peralta L, Forney LJ (2011) Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A 108(Suppl 1):4680–4687. doi:10.1073/pnas.1002611107
Rehman A, Sina C, Gavrilova O, Hasler R, Ott S, Baines JF, Schreiber S, Rosenstiel P (2011) Nod2 is essential for temporal development of intestinal microbial communities. Gut 60(10):1354–1362. doi:10.1136/gut.2010.216259
Rescigno M (2014) Intestinal microbiota and its effects on the immune system. Cell Microbiol. doi:10.1111/cmi.12301
Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI (2013) Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341(6150):1241214. doi:10.1126/science.1241214
Rinttila T, Lyra A, Krogius-Kurikka L, Palva A (2011) Real-time PCR analysis of enteric pathogens from fecal samples of irritable bowel syndrome subjects. Gut Pathog 3(1):6. doi:10.1186/1757-4749-3-6
Rogers CJ, Prabhu KS, Vijay-Kumar M (2014) The microbiome and obesity-an established risk for certain types of cancer. Cancer J 20(3):176–180. doi:10.1097/PPO.0000000000000049
Rogler G (2011) Interaction between susceptibility and environment. Examples from the digestive tract. Dig Dis 29(2):136–143. doi:10.1159/000323876
Rogler G, Holler E (2004) Can NOD2/CARD15 mutations predict intestinal graft-versus-host disease and aid our understanding of Crohn’s disease? Nat Clin Pract Gastroenterol Hepatol 1(2):62–63. doi:10.1038/ncpgasthep0042
Rogler G, Rosano G (2014) The heart and the gut. Eur Heart J 35(7):426–430. doi:10.1093/eurheartj/eht271
Sakata S, Tonooka T, Ishizeki S, Takada M, Sakamoto M, Fukuyama M, Benno Y (2005) Culture-independent analysis of fecal microbiota in infants, with special reference to Bifidobacterium species. FEMS Microbiol Lett 243(2):417–423. doi:10.1016/j.femsle.2005.01.002
Salonen A, de Vos WM (2014) Impact of diet on human intestinal microbiota and health. Annu Rev Food Sci Technol 5:239–262. doi:10.1146/annurev-food-030212-182554
Salonen A, Lahti L, Salojarvi J, Holtrop G, Korpela K, Duncan SH, Date P, Farquharson F, Johnstone AM, Lobley GE, Louis P, Flint HJ, de Vos WM (2014) Impact of diet and individual variation on intestinal microbiota composition and fermentation products in obese men. ISME J. doi:10.1038/ismej.2014.63
Santacruz A, Marcos A, Wärnberg J, Martí A, Martin-Matillas M, Campoy C, Moreno LA, Veiga O, Redondo-Figuero C, Garagorri JM, Azcona C, Delgado M, García-Fuentes M, Collado MC, Sanz Y (2009) Interplay between weight loss and gut microbiota composition in overweight adolescents. Obesity (Silver Spring) 17(10):1906–1915. doi:10.1038/oby.2009.112
Saulnier DM, Riehle K, Mistretta TA, Diaz MA, Mandal D, Raza S, Weidler EM, Qin X, Coarfa C, Milosavljevic A, Petrosino JF, Highlander S, Gibbs R, Lynch SV, Shulman RJ, Versalovic J (2011) Gastrointestinal microbiome signatures of pediatric patients with irritable bowel syndrome. Gastroenterology 141(5):1782–1791. doi:10.1053/j.gastro.2011.06.072
Savage DC (1977) Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol 31:107–133. doi:10.1146/annurev.mi.31.100177.000543
Scanlan PD, Shanahan F, Clune Y, Collins JK, O’Sullivan GC, O’Riordan M, Holmes E, Wang Y, Marchesi JR (2008) Culture-independent analysis of the gut microbiota in colorectal cancer and polyposis. Environ Microbiol 10(3):789–798. doi:10.1111/j.1462-2920.2007.01503.x
Scanlan PD, Shanahan F, O’Mahony C, Marchesi JR (2006) Culture-independent analyses of temporal variation of the dominant fecal microbiota and targeted bacterial subgroups in Crohn’s disease. J Clin Microbiol 44(11):3980–3988. doi:10.1128/JCM. 00312-06
Scharl M, Rogler G (2012) Inflammatory bowel disease pathogenesis. What is new? Curr Opin Gastroenterol 28(4):301–309. doi:10.1097/MOG.0b013e328353e61e
Scher JU, Abramson SB (2011) The microbiome and rheumatoid arthritis. Nat Rev Rheumatol 7(10):569–578. doi:10.1038/nrrheum.2011.121
Schroeder BO, Wu Z, Nuding S, Groscurth S, Marcinowski M, Beisner J, Buchner J, Schaller M, Stange EF, Wehkamp J (2011) Reduction of disulphide bonds unmasks potent antimicrobial activity of human beta-defensin 1. Nature 469(7330):419–423. doi:10.1038/nature09674
Schwabe RF, Jobin C (2013) The microbiome and cancer. Nat Rev Cancer 13(11):800–812. doi:10.1038/nrc3610
Seksik P, Rigottier-Gois L, Gramet G, Sutren M, Pochart P, Marteau P, Jian R, Dore J (2003) Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut 52(2):237–242
Shackel NA, Vadas MA, Gamble JR, McCaughan GW (2014) Beyond liver fibrosis. Hepatic stellate cell senescence links obesity to liver cancer by way of the microbiome. Hepatology 59(6):2413–2415. doi:10.1002/hep.26932
Shaw SY, Blanchard JF, Bernstein CN (2011) Association between the use of antibiotics and new diagnoses of Crohn’s disease and ulcerative colitis. Am J Gastroenterol 106(12):2133–2142. doi:10.1038/ajg.2011.304
Shen W, Gaskins HR, McIntosh MK (2014) Influence of dietary fat on intestinal microbes, inflammation, barrier function and metabolic outcomes. J Nutr Biochem 25(3):270–280. doi:10.1016/j.jnutbio.2013.09.009
Singh S, Nagpal SJS, Murad MH, Yadav S, Kane SV, Pardi DS, Talwalkar JA, Loftus EV (2014) Inflammatory bowel disease is associated with an increased risk of melanoma: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 12(2):210–218
Slack E, Hapfelmeier S, Stecher B, Velykoredko Y, Stoel M, Lawson MA, Geuking MB, Beutler B, Tedder TF, Hardt WD, Bercik P, Verdu EF, McCoy KD, Macpherson AJ (2009) Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science 325(5940):617–620. doi:10.1126/science.1172747
Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, Kau AL, Rich SS, Concannon P, Mychaleckyj JC, Liu J, Houpt E, Li JV, Holmes E, Nicholson J, Knights D, Ursell LK, Knight R, Gordon JI (2013) Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science 339(6119):548–554. doi:10.1126/science.1229000
Stamler J, Rains-Clearman D, Lenz-Litzow K, Tillotson JL, Grandits GA (1997) Relation of smoking at baseline and during trial years 1–6 to food and nutrient intakes and weight in the special intervention and usual care groups in the multiple risk factor intervention trial. Am J Clin Nutr 65(1 Suppl):374S–402S
Stappenbeck TS, Hooper LV, Gordon JI (2002) Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci U S A 99(24):15451–15455. doi:10.1073/pnas.202604299
Stecher B, Macpherson AJ, Hapfelmeier S, Kremer M, Stallmach T, Hardt WD (2005) Comparison of Salmonella enterica serovar Typhimurium colitis in germfree mice and mice pretreated with streptomycin. Infect Immun 73(6):3228–3241. doi:10.1128/IAI. 73.6.3228-3241.2005
Sun L, Nava GM, Stappenbeck TS (2011) Host genetic susceptibility, dysbiosis, and viral triggers in inflammatory bowel disease. Curr Opin Gastroenterol 27(4):321–327. doi:10.1097/MOG.0b013e32834661b4
Svanborg C, Hedlund M, Connell H, Agace W, Duan RD, Nilsson A, Wullt B (1996) Bacterial adherence and mucosal cytokine responses. Receptors and transmembrane signaling. Ann N Y Acad Sci 797:177–190
Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M, Weber J, Hoffmann U, Schreiber S, Dietel M, Lochs H (2002) Mucosal flora in inflammatory bowel disease. Gastroenterology 122(1):44–54
Swidsinski A, Loening-Baucke V, Lochs H, Hale LP (2005) Spatial organization of bacterial flora in normal and inflamed intestine. a fluorescence in situ hybridization study in mice. World J Gastroenterol : WJG 11(8):1131–1140
Swidsinski A, Loening-Baucke V, Herber A (2009) Mucosal flora in Crohn’s disease and ulcerative colitis - an overview. J Physiol Pharmacol : Off J Pol Physiol Soc 60(Suppl 6):61–71
Tanaka S, Kobayashi T, Songjinda P, Tateyama A, Tsubouchi M, Kiyohara C, Shirakawa T, Sonomoto K, Nakayama J (2009) Influence of antibiotic exposure in the early postnatal period on the development of intestinal microbiota. FEMS Immunol Med Microbiol 56(1):80–87. doi:10.1111/j.1574-695X.2009.00553.x
Tilg H (2010) Obesity, metabolic syndrome, and microbiota. multiple interactions. J Clin Gastroenterol 44(Suppl 1):S16–8. doi:10.1097/MCG.0b013e3181dd8b64
Tillisch K, Labus J, Kilpatrick L, Jiang Z, Stains J, Ebrat B, Guyonnet D, Legrain–Raspaud S, Trotin B, Naliboff B, Mayer EA (2013) Consumption of fermented milk product with probiotic modulates brain activity. Gastroenterology 144(7):1394–1401, e4
Tomasini-Johansson BR, Kaufman NR, Ensenberger MG, Ozeri V, Hanski E, Mosher DF (2001) A 49-residue peptide from adhesin F1 of Streptococcus pyogenes inhibits fibronectin matrix assembly. J Biol Chem 276(26):23430–23439. doi:10.1074/jbc.M103467200
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444(7122):1027–1031. doi:10.1038/nature05414
Turner JR (2009) Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 9(11):799–809. doi:10.1038/nri2653
Tuupanen S, Alhopuro P, Mecklin JP, Jarvinen H, Aaltonen LA (2007) No evidence for association of NOD2 R702W and G908R with colorectal cancer. International journal of cancer. J Int Cancer 121(1):76–79. doi:10.1002/ijc.22651
Ursell LK, Haiser HJ, van Treuren W, Garg N, Reddivari L, Vanamala J, Dorrestein PC, Turnbaugh PJ, Knight R (2014) The intestinal metabolome: an intersection between microbiota and host. Gastroenterology 146(6):1470–1476. doi:10.1053/j.gastro.2014.03.001
Vaahtovuo J, Munukka E, Korkeamäki M, Luukkainen R, Toivanen P (2008) Fecal microbiota in early rheumatoid arthritis. J Rheumatol 35(8):1500–1505
Ventura M, Turroni F, Motherway MO, MacSharry J, van Sinderen D (2012) Host-microbe interactions that facilitate gut colonization by commensal bifidobacteria. Trends Microbiol 20(10):467–476. doi:10.1016/j.tim.2012.07.002
Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, Schlitzer A, Ginhoux F, Apetoh L, Chachaty E, Woerther PL, Eberl G, Berard M, Ecobichon C, Clermont D, Bizet C, Gaboriau-Routhiau V, Cerf-Bensussan N, Opolon P, Yessaad N, Vivier E, Ryffel B, Elson CO, Dore J, Kroemer G, Lepage P, Boneca IG, Ghiringhelli F, Zitvogel L (2013) The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342(6161):971–976. doi:10.1126/science.1240537
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT (2010) Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328(5975):228–231. doi:10.1126/science.1179721
Vrieze A, van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, van Hylckama Vlieg JE, Bloks VW, Groen AK, Heilig HG, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JB, Nieuwdorp M (2012) Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143(4):913–6. doi:10.1053/j.gastro.2012.06.031, e7
Walter J, Ley R (2011) The human gut microbiome: ecology and recent evolutionary changes. Annu Rev Microbiol 65:411–429. doi:10.1146/annurev-micro-090110-102830
Wang L, Conlon MA, Christophersen CT, Sorich MJ, Angley MT (2014) Gastrointestinal microbiota and metabolite biomarkers in children with autism spectrum disorders. Biomark Med 8(3):331–344. doi:10.2217/bmm.14.12
Wang P, Zhang L, Jiang JM, Ma D, Tao HX, Yuan SL, Wang YC, Wang LC, Liang H, Zhang ZS, Liu CJ (2012) Association of NOD1 and NOD2 genes polymorphisms with Helicobacter pylori related gastric cancer in a Chinese population. World J Gastroenterol : WJG 18(17):2112–2120. doi:10.3748/wjg.v18.i17.2112
Ward ME, Watt PJ (1972) Adherence of Neisseria gonorrhoeae to urethral mucosal cells: an electron-microscopic study of human gonorrhea. J Infect Dis 126(6):601–605
Watson AJ, Chu S, Sieck L, Gerasimenko O, Bullen T, Campbell F, McKenna M, Rose T, Montrose MH (2005) Epithelial barrier function in vivo is sustained despite gaps in epithelial layers. Gastroenterology 129(3):902–912. doi:10.1053/j.gastro.2005.06.015
Watson AJ, Hughes KR (2012) TNF-alpha-induced intestinal epithelial cell shedding: implications for intestinal barrier function. Ann N Y Acad Sci 1258:1–8. doi:10.1111/j.1749-6632.2012.06523.x
Wehkamp J, Koslowski M, Wang G, Stange EF (2008) Barrier dysfunction due to distinct defensin deficiencies in small intestinal and colonic Crohn’s disease. Mucosal Immunol 1(Suppl 1):S67–74. doi:10.1038/mi.2008.48
Wehkamp J, Stange EF (2006) A new look at Crohn’s disease: breakdown of the mucosal antibacterial defense. Ann N Y Acad Sci 1072:321–331. doi:10.1196/annals.1326.030
Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV (2008) Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455(7216):1109–1113. doi:10.1038/nature07336
Wijburg OL, Uren TK, Simpfendorfer K, Johansen FE, Brandtzaeg P, Strugnell RA (2006) Innate secretory antibodies protect against natural Salmonella typhimurium infection. J Exp Med 203(1):21–26. doi:10.1084/jem.20052093
Wold AE, Thorssen M, Hull S, Eden CS (1988) Attachment of Escherichia coli via mannose- or Gal alpha 1–4Gal beta-containing receptors to human colonic epithelial cells. Infect Immun 56(10):2531–2537
Wood NJ (2012) Microbiota: dysbiosis driven by inflammasome deficiency exacerbates hepatic steatosis and governs rate of NAFLD progression. Nat Rev Gastroenterol Hepatol 9(3):123. doi:10.1038/nrgastro.2012.21
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI (2012) Human gut microbiome viewed across age and geography. Nature 486(7402):222–227. doi:10.1038/nature11053
Young D, Hussell T, Dougan G (2002) Chronic bacterial infections: living with unwanted guests. Nat Immunol 3(11):1026–1032. doi:10.1038/ni1102-1026
Zoetendal EG, de Vos WM (2014) Effect of diet on the intestinal microbiota and its activity. Curr Opin Gastroenterol 30(2):189–195. doi:10.1097/MOG.0000000000000048
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Beat Steinmann
Rights and permissions
About this article

Cite this article
Biedermann, L., Rogler, G. The intestinal microbiota: its role in health and disease. Eur J Pediatr 174, 151–167 (2015). https://doi.org/10.1007/s00431-014-2476-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-014-2476-2