
Abstract
Infantile haemangioma (IH) is the most frequent childhood tumour. Although it is benign and self-limiting, severe complications can arise due to localisation and fast tumour growth. Management and therapy of IH has changed greatly after 2008 with propranolol. However, the pathogenesis remains elusive. This update provides an overview of all possible mechanisms currently considered. We discuss the possibility that several mechanisms act together, although local hypoxia seems to be important. Clinically, in about half of the cases, an IH is preceded by an anaemic macula (local ischaemia) or a so-called precursor lesion. Laboratory findings indicate stabilisation and an increased transcription activity of hypoxia-inducible factor 1 alpha (HIF1α), leading to up-regulation of its downstream target genes (such as vascular endothelial growth factor (VEGF)), which normally occurs in cases of hypoxia.
Conclusion: Three main hypotheses have been proposed, namely (1) the theory of tissue hypoxia, (2) the theory of embolization of placental endothelial cells and (3) the theory of increased angiogenic and vasculogenic activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
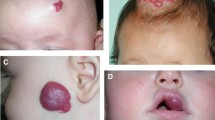
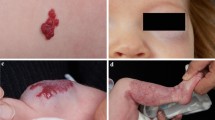
Reported incidences of infantile haemangioma (IH) vary greatly, but in any case, it is the most frequent childhood tumour, with incidences of 5–10 % up to 20 % in prematurely born infants [11, 15, 12]. These tumours occur predominantly in the Caucasian population [37]. IHs follow a typical course: they arise within the first few days to weeks after birth and most IH grow exponentially for up to 6 to 9 months. Hereafter, regression follows, by approximately 10 % per year [34]. Thus, most IHs have gone at the age of 10 years but a scar can remain [4]. Although IHs are benign and self-limiting, severe complications may arise due to localisation and accelerated tumour growth [22]. Figures 1, 2, 3, 4, 5, 6 and 7 show several different IHs. Despite extensive literature (especially after 2008), the pathogenesis is still not clear [46]. This update provides an overview of mechanisms that are currently being considered, and some aspects of these theories are discussed.

Precursor lesion before development of an infantile haemangioma

Infantile haemangioma in the active (proliferative) phase

Infantile haemangioma with both deep swelling and a superficial component

Alarming infantile haemangioma: risk for eye abnormalities

An infantile haemangioma with ulceration and great risk of permanent deformity

Infantile haemangioma in the involutive phase

Haemangiomatosis
In a proliferative IH, rapidly growing endothelial cells form blood vessels. Increased apoptosis of endothelial cells in the involution phase leads to regression of blood vessels. Eventually, the thick multilaminated basement membrane surrounding the endothelial layer is replaced by adipocytes in fibrous tissue [9]. Furthermore, a considerable increase in the number of mast cells during the involution phase may alter the balance of angiogenic factors, thus promoting regression [44]. The empirically based current therapy aims to induce/accelerate the natural involution process [9]. Systemic propranolol induces quick therapeutic responses and has made corticosteroids and all other treatment options obsolete [29, 49]. However, propranolol has potential side effects and sometimes must be used for 1–2 years (own experience). As topical use has limitations as well, we are still searching for a better alternative. Propranolol and corticosteroids both act on factors induced by hypoxia-inducible factor 1 alpha (HIF1α) as a result of local hypoxia [20, 49], supporting a crucial role of local hypoxia in IH. A better understanding of the pathogenesis may improve targeted therapy options in the management of IH. The therapy of IH will be discussed in part II which will appear in the next issue [25].
Characteristics of proliferating versus involuting IHs [30]
IHs consist of multipotent stem cells (CD133+), immature endothelial cells (CD31+), pericytes (SMA+), dendritic cells (factor XIIIa+) and mesenchymal cells (with adipogenic potential). During the proliferative phase, endothelial and interstitial cells express a marker of proliferation, namely the antibody MIB-1. Furthermore, CD31+ endothelial cells are clonal and express a particular phenotype: indoleamine 2,3-dioxygenase (IDO), LYVE-1, merosin, CCR6, glucose transporter 1 (GLUT-1), antigen Lewis Y (Ley), antigen FcγRII, and CD15. This phenotype changes over time with the maturation of endothelial cells (see Fig. 8). However, GLUT-1 stays positive and therefore can discriminate between IHs and other vascular malformations [32, 39]. During involution, endothelial cells express caspases, which are known markers of apoptosis. Simultaneously, there is an increase in the expression of markers of maturation and activation of endothelial cells such as HLA-DR and ICAM-1 (CD54). Mesenchymal cells differentiate into adipocytes at this stage. Moreover, in a recent report, it was concluded that apoptosis is prevented in proliferative IHs by an up-regulated autocrine vascular endothelial growth factor (VEGF)/VEGF receptor 2 (VEGFR2) signalling loop. VEGF also activates the survival-promoting PI3K/Akt pathway. Activation of Akt in turn stimulates the expression of anti-apoptotic proteins, such as Bcl-2. Thus, the up-regulated autocrine VEGF loop promotes IH-derived endothelial cells survival via regulation of the PI3K/Akt/Bcl-2 pathway [26].

Simplification of the interaction between the hypoxia-inducible factor (HIF) pathway, the mammalian target of rapamycin (mTOR) pathway and several factors, resulting from local hypoxia into endothelial cell proliferation (proliferative infantile haemangioma). Explanation: hypoxia triggers stabilisation at the protein level of the transcription factor hypoxia-inducible factor 1 alpha (HIF1α). HIF1α in turn stimulates transcription of downstream target genes such as those encoding BNIP3, CA-IX, glucose transporter 1 (GLUT-1), phosphorylated protein kinase B (pAKT), phosphorylated S6 protein (pS6) and vascular endothelial growth factor (VEGF). These target genes can be regulated either directly by HIF1α or by hypoxia-induced regulation of mammalian target of rapamycin complex 1 (mTORC1) signalling. mTORC1 is a key player in the mTOR pathway, a protein complex with a central role in regulating cellular metabolism, driven by growth factors and nutrients as well as hypoxia
Hypotheses
Many mechanisms have been considered to explain the development of IHs. Three competing hypotheses are currently being considered, which are, however, not mutually exclusive [23]:
-
1.
Tissue hypoxia
-
2.
Embolization of placental endothelial cells
-
3.
Increased angiogenic and vasculogenic activity.
Tissue hypoxia seems to be the most powerful inducer of angiogenesis (and vasculogenesis). A relation was found between placental hypoxia and IHs [16]. Also, the relationship between low birth weight and IH and the association between ROP and IH points to hypoxia [17].
As a less important hypothesis, genetic involvement has been proposed [51].
Hypoxia
Local hypoxia may be involved in the pathogenesis of IH [5, 14, 28, 33]. In 50 % of cases, the skin is blanched (precursor lesion) at the site where an IH will eventually develop, supporting the idea that local ischaemia is important. A hypoxic environment triggers stabilisation at the protein level of the transcription factor HIF1α [52]. HIF1α in turn stimulates transcription of downstream target genes such as Bcl-2/adenovirus E1B kilodalton-interacting protein (BNIP) family member 3 (BNIP3), carbon anhydrase IX (CA-IX), GLUT-1, pAKT, pS6 and VEGF [6]. These target genes might be regulated either directly by HIF1α signalling or by hypoxia-induced regulation of mammalian target of rapamycin complex 1 (mTORC1) signalling [1]. mTORC1 is a key player in the mTOR pathway, a protein complex with a central role in regulating cellular metabolism, driven by growth factors, nutrients as well as hypoxia (Fig. 9). Deregulation of the mTOR pathway may lead to disorganised growth [53]. As macrophages secrete pro-angiogenic molecules such as TNF-α and interleukin-1, they are also thought to be involved in the evolution of IHs [9, 28, 43]. Of all theories proposed, the hypoxia theory seems to be attractive, given the anaemic macula (precursor lesion) often seen and the endothelial cell origin of IHs (cells typically growing under hypoxic conditions) [24]. It is known that the target genes (VEGF, GLUT-1, etc.) can also be stimulated by hypoxia via hypoxia-inducible factor 2 alpha (HIF2α) (alone or in combination with HIF1α), with the same result [18, 42, 45]. HIF1 is a heterodimer of two proteins: HIF1α and HIF1β. HIF2α forms a functional heterodimer with HIF1β, resulting in the HIF2 complex, which activates transcription from the same DNA recognition sites as HIF1. This activation is stimulated under hypoxic conditions [18, 42, 45]. Circulating bone marrow-derived endothelial progenitor cells form new blood vessels in ischemic tissues using mediators regulated by HIF1α. Mobilization is enhanced by VEGF, MMP9 and oestrogen, whereas homing is secondary to localized expression of SDF1α [28].

Pathophysiological mechanisms at a cellular level in the course of infantile haemangiomas: a stem cell (CD133), under hypoxic conditions resulting in the activation of the HIF pathway and overexpression of VEGF, multiplicates and differentiates into endothelial progenitor cells (CD31+), mesenchymal cell precursors of adipocytes and pericytes. Based on Fig. 6 of the article of Léauté-Labrèze et al. [30]
Placental origin
The placental theory is attractive because it would explain the programmed life cycle of IH. IH might represent benign metastases originating from the placenta or other cells that proliferate in areas of low oxygen tension, such as the “end artery, vascular dead end” sites occurring in embryonic fusion planes [36]. Therefore, placental embolization is thought to play a causative role [2, 50]. Chorionic villus sampling has been associated with an increased incidence of IHs [13]. GLUT-1 is strongly expressed in IHs, but not in other vascular malformations; GLUT-1 is also expressed in the placenta. Furthermore, IHs and the placenta also express other molecular markers such as merosin, laminin, Lewis Y antigen, FcγRII, IDO and IGF-2 [40, 41]. It has also been noted that placenta and IH have high levels of genetic similarity when compared with other vascular tumours and normal structures [3]. Therefore, it has been hypothesized that IH precursor cells originate from the placenta, although subsequent molecular genetic investigations revealed no evidence for maternal-foetal microchimerism [23]. This, however, does not rule out the possibility of the placental origin of IH tissue because the placenta is predominantly foetal in origin.
Increased angiogenic and vasculogenic activity
Vasculogenesis versus angiogenesis [19]
Both vasculogenesis and angiogenesis have been proposed as mechanisms contributing to the neovascularization in IH. Vasculogenesis is the de novo formation of blood vessels from stem cells. It was long believed that this occurs in foetal life only. Angiogenesis on the other hand is the growth of new blood vessels from pre-existing vessels, which includes migration of endothelial cells.
The group of Greenberger found in 2008 that mesenchymal cells, isolated from proliferative IHs using CD133-coated magnetic beads, are capable of differentiating into endothelial cells, pericytes (perivascular cells) and adipogenic lineages [19]. When implanted into immune-deficient mice, these IH-derived stem cells formed GLUT-1-positive vessels. Greenberger et al. [20, 21] therefore concluded that vasculogenesis is an important mechanism underlying IH genesis. Khan et al. found evidence that CD133-selected IH-derived stem cells recapitulate human IH in a murine in vivo model [27]. This did not work with IH-derived endothelial cells. Clonal IH-derived stem cells produced human GLUT-1-positive microvessels and, after a while, also human adipocytes. These results demonstrate that IH-derived stem cells are the cellular precursors of IHs. Similar results were found by Xu et al. [54].
In the proliferative phase, the blood vessels are small and the endothelium is plump and metabolically active, suggesting an immature phenotype. Mulliken et al. have shown that IH-derived endothelial cells form capillary-like tubes in vitro [38]. Boye et al. showed that IH-derived endothelial cells are clonal and therefore suggested that they arise from a common precursor [10]. First, IH-derived stem cells differentiate into endothelial cells due to (local) hypoxia (vasculogenesis). Because of juxtacrine signalling between IH-derived endothelial cells and IH-derived stem cells via Jagged1 signalling through the Notch pathway, IH-derived stem cells differentiate into pericytes [8]. There are many pericytes in the proliferating phase, and they appear to undergo a maturation process concurrently with the endothelial cells. Recently, it was found that pericytes in IH are pro-angiogenic [7]. This triggers angiogenesis.
Other factors
E-selectin, normally found in inflammatory skin, can also be found in proliferating IHs and its expression decreases in involuting IHs [48]. In another study, Smadja et al. found evidence that α6-integrin is increased in proliferating IHs and expressed by IH-derived stem cells [47]. This expression is decreased in involuting IHs. Integrins are receptors important for cellular adhesion to extracellular matrix and to other cells. Furthermore, α6-integrin is also involved in angiogenesis and is required to form vascular networks in vitro. Finally, hormonal influences may be involved. Oestrogen receptors are also expressed by IH-derived endothelial cells, and stimulation with oestrogen increases proliferation, migration and survival of endothelial cells [9, 28]. Genetic influences may contribute as several patients with IHs show a considerable loss of heterozygosity for markers in a region of chromosome 5q [31, 51]. The evidence, however, is not conclusive and could not be confirmed in bigger studies.
Treatment based on pathogenesis: rapamycin [21, 35]
Treatment, if necessary, is usually with propranolol nowadays. Before 2008, corticosteroids were the traditional first-line therapy. Pointing at side effects and non-responders to therapy (especially in the case of corticosteroids), Greenberger et al. make a plea for additional therapies that will shorten treatment duration or may even prevent problematic IHs from forming [21]. In their murine model, they tested rapamycin, which is an inhibitor of the mTOR pathway. They concluded that rapamycin suppresses vasculogenesis in vivo, that self-renewal and multi-lineage differentiation are disrupted by rapamycin, that rapamycin leads to mesenchymal maturation and impaired vasculogenic potential, and that rapamycin stimulates regression of pre-existing vessels formed from IH-derived stem cells. Another option is the monoclonal antibody bevacizumab, which, however, has never been tested in IH [9].
Overall conclusion
The pathogenesis of infantile haemangioma remains elusive. There are currently three competing hypotheses which are, however, not mutually exclusive: (1) the theory of tissue hypoxia, (2) the theory of embolization of placental endothelial cells and (3) the theory of increased angiogenic and vasculogenic activity. Local hypoxia is important: laboratory findings indicate stabilisation and an increased transcription activity of hypoxia-inducible factor 1 alpha (HIF1α), leading to up-regulation of its downstream target genes (such as vascular endothelial growth factor (VEGF)), which normally occurs in cases of hypoxia.
Abbreviations
- BNIP3:
-
Bcl-2/adenovirus E1B kilodalton-interacting protein (BNIP) family member 3
- CA-IX:
-
Carbon anhydrase IX
- GLUT-1:
-
Glucose transporter 1
- HIF1α:
-
Hypoxia-inducible factor 1 alpha
- HIF2α:
-
Hypoxia-inducible factor 2 alpha
- IDO:
-
Indoleamine 2,3-dioxygenase
- IGF:
-
Insulin-like growth factor
- IH:
-
Infantile haemangioma
- MMP9:
-
Matrix metallopeptidase 9
- mTOR:
-
Mammalian target of rapamycin
- mTORC1:
-
mTOR complex 1
- pAKT:
-
Phosphorylated v-akt murine thymoma viral oncogene homolog 1
- pS6:
-
Phosphorylated S6 protein
- ROP:
-
Retinopathy of prematurity
- SDF1α:
-
Stromal cell-derived factor 1 alpha
- TNF-α:
-
Tumour necrosis factor alpha
- VEGF:
-
Vascular endothelial growth factor
- VEGF-A:
-
Vascular endothelial growth factor A
- VEGFR:
-
Vascular endothelial growth factor receptor
References
Arsham AM, Howell JJ, Simon MC (2003) A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem 278(32):29655–29660
Barnes CM, Christison-Lagay EA, Folkman J (2007) The placenta theory and the origin of infantile hemangioma. Lymphat Res Biol 5(4):245–255
Barnes CM, Huang S, Kaipainen A, Sanoudou D, Chen EJ, Eichler GS, Guo Y, Yu Y, Ingber DE, Mulliken JB, Beggs AH, Folkman J, Fishman SJ (2005) Evidence by molecular profiling for a placental origin of infantile hemangioma. Proc Natl Acad Sci U S A 102(52):19097–19102
Bauland CG, Luning TH, Smit JM, Zeebregts CJ, Spauwen PH (2011) Untreated hemangiomas: growth pattern and residual lesions. Plast Reconstr Surg 127(4):1643–1648
Bauland CG, van Steensel MA, Steijlen PM, Rieu PN, Spauwen PH (2006) The pathogenesis of hemangiomas: a review. Plast Reconstr Surg 117(2):29e–35e
Boscolo E, Mulliken JB, Bischoff J (2011) VEGFR-1 mediates endothelial differentiation and formation of blood vessels in a murine model of infantile hemangioma. Am J Pathol 179(5):2266–2277
Boscolo E, Mulliken JB, Bischoff J (2013) Pericytes from infantile hemangioma display proangiogenic properties and dysregulated angiopoietin-1. Arterioscler Thromb Vasc Biol 33(3):501–509
Boscolo E, Stewart CL, Greenberger S, Wu JK, Durham JT, Herman IM, Mulliken JB, Kitajewski J, Bischoff J (2011) JAGGED1 signaling regulates hemangioma stem cell-to-pericyte/vascular smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol 31(10):2181–2192
Boye E, Jinnin M, Olsen BR (2009) Infantile hemangioma: challenges, new insights, and therapeutic promise. J Craniofac Surg 20(Suppl 1):678–684
Boye E, Yu Y, Paranya G, Mulliken JB, Olsen BR, Bischoff J (2001) Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest 107(6):745–752
Bruckner AL, Frieden IJ (2003) Hemangiomas of infancy. J Am Acad Dermatol 48(4):477–493, quiz 494-476
Bruckner AL, Frieden IJ (2006) Infantile hemangiomas. J Am Acad Dermatol 55(4):671–682
Burton BK, Schulz CJ, Angle B, Burd LI (1995) An increased incidence of haemangiomas in infants born following chorionic villus sampling (CVS). Prenat Diagn 15(3):209–214
Chang EI, Chang EI, Thangarajah H, Hamou C, Gurtner GC (2007) Hypoxia, hormones, and endothelial progenitor cells in hemangioma. Lymphat Res Biol 5(4):237–243
Chiller KG, Passaro D, Frieden IJ (2002) Hemangiomas of infancy: clinical characteristics, morphologic subtypes, and their relationship to race, ethnicity, and sex. Arch Dermatol 138(12):1567–1576
Colonna V, Resta L, Napoli A, Bonifazi E (2009) Placental hypoxia and neonatal haemangioma: clinical and histological observations. Br J Dermatol 162(1):208–9
Drolet BA, Frieden IJ (2010) Characteristics of infantile hemangiomas as clues to pathogenesis: does hypoxia connect the dots? Arch Dermatol 146(11):1295–1299
Giatromanolaki A, Arvanitidou V, Hatzimichael A, Simopoulos C, Sivridis E (2005) The HIF-2alpha/VEGF pathway activation in cutaneous capillary haemangiomas. Pathology 37(2):149–151
Greenberger S, Bischoff J (2013) Pathogenesis of infantile haemangioma. Br J Dermatol 169(1):12–19
Greenberger S, Boscolo E, Adini I, Mulliken JB, Bischoff J (2010) Corticosteroid suppression of VEGF-A in infantile hemangioma-derived stem cells. N Engl J Med 362(11):1005–1013
Greenberger S, Yuan S, Walsh LA, Boscolo E, Kang KT, Matthews B, Mulliken JB, Bischoff J (2011) Rapamycin suppresses self-renewal and vasculogenic potential of stem cells isolated from infantile hemangioma. J Invest Dermatol 131(12):2467–2476
Haggstrom AN, Drolet BA, Baselga E, Chamlin SL, Garzon MC, Horii KA, Lucky AW, Mancini AJ, Metry DW, Newell B, Nopper AJ, Frieden IJ (2006) Prospective study of infantile hemangiomas: clinical characteristics predicting complications and treatment. Pediatrics 118(3):882–887
Hoeger PH (2011) Infantile haemangioma: new aspects on the pathogenesis of the most common skin tumour in children. Br J Dermatol 164(2):234–235
Janmohamed SR, Brinkhuizen T, Madern GC, den Hollander JC, de Laat PC, Van Steensel MA, Oranje AP (2014) Support for the hypoxia theory in the pathogenesis of infantile haemangioma. Clin Exp Dermatol. (in press)
Janmohamed SR, Madern GC, de Laat PC, Oranje AP (2014) Educational paper: therapy of infantile hemangioma—history and current state (part II). Eur J Pediatr. doi:10.1007/s00431-014-2404-5
Ji Y, Chen S, Li K, Xiao X, Xu T, Zheng S (2014) Upregulated autocrine vascular endothelial growth factor (VEGF)/VEGF receptor-2 loop prevents apoptosis in haemangioma-derived endothelial cells. Br J Dermatol 170(1):78–86
Khan ZA, Boscolo E, Picard A, Psutka S, Melero-Martin JM, Bartch TC, Mulliken JB, Bischoff J (2008) Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest 118(7):2592–2599
Kleinman ME, Greives MR, Churgin SS, Blechman KM, Chang EI, Ceradini DJ, Tepper OM, Gurtner GC (2007) Hypoxia-induced mediators of stem/progenitor cell trafficking are increased in children with hemangioma. Arterioscler Thromb Vasc Biol 27(12):2664–2670
Leaute-Labreze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taieb A (2008) Propranolol for severe hemangiomas of infancy. N Engl J Med 358(24):2649–2651
Léauté-Labrèze C, Prey S, Ezzedine K (2011) Infantile haemangioma: part I. Pathophysiology, epidemiology, clinical features, life cycle and associated structural abnormalities. J Eur Acad Dermatol Venereol 25(11):1245–1253
Lee KC, Bercovitch L (2013) Update on infantile hemangiomas. Semin Perinatol 37(1):49–58
Leon-Villapalos J, Wolfe K, Kangesu L (2005) GLUT-1: an extra diagnostic tool to differentiate between haemangiomas and vascular malformations. Br J Plast Surg 58(3):348–352
Lo K, Mihm M, Fay A (2009) Current theories on the pathogenesis of infantile hemangioma. Semin Ophthalmol 24(3):172–177
Luu M, Frieden IJ (2013) Haemangioma: clinical course, complications and management. Br J Dermatol 169(1):20–30
Medici D, Olsen BR (2012) Rapamycin inhibits proliferation of hemangioma endothelial cells by reducing HIF-1-dependent expression of VEGF. PLoS One 7(8):e42913
Mihm MC Jr, Nelson JS (2010) Hypothesis: the metastatic niche theory can elucidate infantile hemangioma development. J Cutan Pathol 37(Suppl 1):83–87
Mulliken JB, Enjolras O (2004) Congenital hemangiomas and infantile hemangioma: missing links. J Am Acad Dermatol 50(6):875–882
Mulliken JB, Zetter BR, Folkman J (1982) In vitro characteristics of endothelium from hemangiomas and vascular malformations. Surgery 92(2):348–353
North PE, Waner M, Mizeracki A, Mihm MC Jr (2000) GLUT1: a newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol 31(1):11–22
North PE, Waner M, Mizeracki A, Mrak RE, Nicholas R, Kincannon J, Suen JY, Mihm MC Jr (2001) A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol 137(5):559–570
Phung TL, Hochman M (2012) Pathogenesis of infantile hemangioma. Facial Plast Surg 28(6):554–562
Rankin EB, Rha J, Unger TL, Wu CH, Shutt HP, Johnson RS, Simon MC, Keith B, Haase VH (2008) Hypoxia-inducible factor-2 regulates vascular tumorigenesis in mice. Oncogene 27(40):5354–5358
Ritter MR, Butschek RA, Friedlander M, Friedlander SF (2007) Pathogenesis of infantile haemangioma: new molecular and cellular insights. Expert Rev Mol Med 9(32):1–19
Ritter MR, Reinisch J, Friedlander SF, Friedlander M (2006) Myeloid cells in infantile hemangioma. Am J Pathol 168(2):621–628
Shinojima T, Oya M, Takayanagi A, Mizuno R, Shimizu N, Murai M (2007) Renal cancer cells lacking hypoxia inducible factor (HIF)-1alpha expression maintain vascular endothelial growth factor expression through HIF-2alpha. Carcinogenesis 28(3):529–536
Sidbury R (2010) Update on vascular tumors of infancy. Curr Opin Pediatr 22(4):432–437
Smadja DM, Guerin CL, Boscolo E, Bieche I, Mulliken JB, Bischoff J (2013) α6-Integrin is required for the adhesion and vasculogenic potential of hemangioma stem cells. Stem Cells 32(3):684–93
Smadja DM, Mulliken JB, Bischoff J (2012) E-selectin mediates stem cell adhesion and formation of blood vessels in a murine model of infantile hemangioma. Am J Pathol 181(6):2239–2247
Storch CH, Hoeger PH (2010) Propranolol for infantile haemangiomas: insights into the molecular mechanisms of action. Br J Dermatol 163(2):269–274
Sun ZY, Yi CG, Zhao H, Yin GQ, Gao M, Liu YB, Qin JD, Wang SF, Guo SZ (2008) Infantile hemangioma is originated from placental trophoblast, fact or fiction? Med Hypotheses 71(3):444–448
Walter JW, Blei F, Anderson JL, Orlow SJ, Speer MC, Marchuk DA (1999) Genetic mapping of a novel familial form of infantile hemangioma. Am J Med Genet 82(1):77–83
Wang GL, Jiang BH, Rue EA, Semenza GL (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 92(12):5510–5514
Wouters BG, Koritzinsky M (2008) Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 8(11):851–864
Xu D, O TM, Shartava A, Fowles TC, Yang J, Fink LM, Ward DC, Mihm MC, Waner M, Ma Y (2011) Isolation, characterization, and in vitro propagation of infantile hemangioma stem cells and an in vivo mouse model. J Hematol Oncol 4:54
Acknowledgments
We thank Ko Hagoort for language revision. This study was funded by project Aardbeesie (www.aardbeesie.nl) and the Foundation for Paediatric Dermatology Rotterdam.
Conflict of interest
The authors declare that they have no conflict of interest
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Beat Steinmann
Rights and permissions
About this article

Cite this article
Janmohamed, S.R., Madern, G.C., de Laat, P.C.J. et al. Educational paper: pathogenesis of infantile haemangioma, an update 2014 (part I). Eur J Pediatr 174, 97–103 (2015). https://doi.org/10.1007/s00431-014-2403-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-014-2403-6