
Abstract
This study was undertaken to investigate the evolution of clinical features between onset of symptoms and diagnosis in children with brain tumours and to identify ways of shortening the time to diagnosis. One hundred and thirty-nine children with a brain tumour were recruited from four UK paediatric neuro-oncology centres. Children had a median of one symptom or sign at symptom onset and six by diagnosis. The symptoms and/or signs experienced at symptom onset and at diagnosis were as follows: headache in 55 and 81 children, nausea and vomiting in 39 and 88 children, motor system abnormalities in 31 and 93 children, cranial nerve palsies in 24 and 75 children, visual system abnormalities in 23 and 96 children, endocrine or growth abnormalities in 10 and 35 children and behavioural change in 4 and 55 children. The median time between symptom onset and diagnosis (symptom interval) was 3.3 months. A longer symptom interval was associated with head tilt, cranial nerve palsies, endocrine and growth abnormalities and reduced visual acuity. More than half of children with brain tumours developed problems with vision and more than a third developed motor problems, cranial nerve palsies, behavioural change, or nausea and vomiting between symptom onset and diagnosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Brain tumours are the commonest solid tumours in children and are the commonest cause of death from cancer in childhood [19, 24, 25]. Despite advances in neuro-imaging, their timely diagnosis remains problematic. Tumour presentation is varied and dependent upon tumour location and age of the child; symptoms may fluctuate in severity [10, 27, 28]. The consequence is that children have often been symptomatic for months before diagnosis is made, frequently in the setting of emergency referral [9, 23].
We undertook a retrospective cohort study of symptom progression in children newly diagnosed with a brain tumour in four paediatric neuro-oncology centres in order to provide contemporary information on their presentation and diagnosis in the UK and on the evolution of clinical features between onset of symptoms and diagnosis. This was the initial stage in a project devising guidance to help healthcare professionals to better identify children who need fast-track imaging for a possible brain tumour.
Methods
Information was obtained from the hospital medical records of children diagnosed with a brain tumour at Birmingham Children's Hospital, Queen's Medical Centre, Nottingham, Southampton General Hospital and Sheffield Children's Hospital between January 2004 and March 2006. Data were collected on the patients' age, sex, ethnic origin, symptom interval, signs and symptoms at disease onset and at diagnosis, deprivation score and healthcare professionals consulted during the symptom interval. Signs and symptoms were recorded as described in the records and then grouped into the following categories: headache, nausea and vomiting, seizures, alteration in or loss of consciousness (excluding seizures), motor system abnormalities (abnormal gait, abnormal coordination, focal motor weakness, involuntary movements, abnormal tone, hemiplegia, paraplegia, quadriplegia, abnormal reflexes, abnormal speech, abnormal handwriting and dystonia), visual system abnormalities (reduced visual acuity, reduced visual fields, nystagmus, other abnormal eye movements, squint, exophthalmia, diplopia, eye pain, papilloedema, optic atrophy, unequal pupils and sunsetting), cranial nerve palsies, abdominal or back pain, spinal deformity, behavioural change (including lethargy and school difficulties), endocrine and growth abnormalities and other findings. Patients' deprivation score was determined using the Index of Multiple Deprivation Score for wards from the Office of National Statistics [12].
Statistical analysis
All analyses were undertaken using SPSS 12.0. Subgroup comparison was undertaken using the Mann–Whitney and Kruskal–Wallis tests. Cox regression analysis was undertaken to explore the relationship between symptom interval and initial sign or symptom and between symptom interval and deprivation score. Fisher's exact test was used to explore the relationship between long (greater than the median) and short (less than or equal to the median) symptom interval and signs and symptoms with unknown date of onset.
Ethics
Approval was granted by Nottingham 2 REC. Written informed consent was provided by patients aged 16 years and above and by the parents or guardians of younger patients.
Results
Patient characteristics
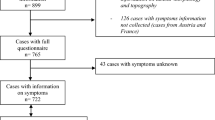
Of 182 children and adolescents diagnosed with a brain tumour at the participating centres during the recruitment period, 139 children were recruited to the study. The median age at diagnosis was 8.1 years (range 29 days to 16.7 years), the male to female ratio was 1.4:1 (82 male, 57 female) and there was a wide range of histological diagnoses (Table 1). An asymptomatic child with tuberous sclerosis was diagnosed with a subependymal giant cell astrocytoma as a result of screening. One tumour (a cerebellar pilocytic astrocytoma) was diagnosed as an incidental finding following imaging to investigate unrelated precocious puberty.
Signs and symptoms
The signs and symptoms at symptom onset, ranked in order of decreasing frequency, were headache, nausea and/or vomiting, motor system abnormalities, cranial nerve palsies, visual system abnormalities, seizures, endocrine or growth abnormalities, behavioural change, abdominal or back pain, an alteration in or loss of consciousness and spinal deformity (Table 2). The most common motor abnormalities seen were abnormalities of gait and coordination, and the commonest visual abnormalities were squint and reduced visual acuity. Of 24 patients with a cranial nerve abnormality at symptom onset, 16 had abnormalities involving the visual system. Lethargy was the only behavioural change identified at symptom onset.
Of 79 children with a single clinical feature at symptom onset, 26 children had a headache, 11 had a visual system abnormality, 10 nausea and/or vomiting, 10 a motor system abnormality, 8 seizures and 4 an endocrine or growth abnormality. Two children had a cranial nerve abnormality not involving the visual system (one hearing loss and one dysphagia).
There was a clear increase in the number of symptoms and signs between symptom onset and diagnosis from a median (range) of 1 (1–8) to 6 (1–16). More than half of the children developed visual system abnormalities and between one third and one half of children developed motor system abnormalities, cranial nerve palsies, behavioural change, and nausea and vomiting between symptom onset and diagnosis (Tables 2 and 3). By the time of diagnosis, 26 children had lost weight and the relative frequency of clinical features had changed with the commonest being visual system abnormalities, seen in 97 patients (Tables 2 and 3). Of 75 children with a cranial nerve abnormality at diagnosis, 48 had an abnormality involving the visual system.
By diagnosis 132 children had signs and symptoms in one or more of the following categories: headache, nausea or vomiting, visual system abnormalities and motor system abnormalities. Only 7 children did not present with symptoms and signs in these categories. Of these, two presented with partial seizures, two with polyuria and polydipsia, one with hearing loss, and two were diagnosed with asymptomatic tumours whilst undergoing investigation of tuberous sclerosis and precocious puberty respectively. By diagnosis, no child had only headache or vomiting, only three children still had only one sign or symptom (one polyuria and polydispsia, one seizures, one hearing loss) and only five children had two signs or symptoms (six motor abnormalities, one headache, one vomiting, one visual abnormality and one growth abnormality).
In contrast to older children, those aged less than 4 years had motor and visual system abnormalities, nausea and vomiting and cranial nerve palsies as the commonest clinical features, both at symptom onset and at diagnosis, while headache was rare and even by diagnosis was a recognised symptom in only eight children (Fig. 1a–c). For children aged less than 4 years, the greatest increase in number of signs and symptoms occurred with motor system abnormalities and behavioural change (Fig. 1a–c). Significant differences between those aged <4, 4–12 and >12 years were seen with respect to frequency of headache, both at symptom onset (p ≤ 0.001) and at diagnosis (p ≤ 0.001), frequency of motor system abnormalities, both at symptom onset (p = 0.04) and at diagnosis (p = 0.02), and frequency of nausea and vomiting at diagnosis (p = 0.01).

Numbers of children showing specific signs and symptoms at symptom onset (grey) and diagnosis (black). a Children aged under 4 years (n = 42). b Children aged 4–12 years (n = 55). c Children aged over 12 years (n = 42). N/V nausea and/or vomiting, Vision visual system abnormality, Motor motor system abnormality, CNP cranial nerve palsy, E/G endocrine or growth abnormality, A/B abdominal or back pain, Consciousness alteration in or loss of consciousness
Symptom interval
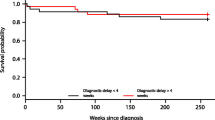
The symptom interval ranged from 0 to 6.9 years (median 3.3 months). Univariate analysis revealed no association between symptom interval and either tumour location, patient age, sex, ethnic origin or deprivation score. High-grade tumours (tumour grading was possible for 119 patients) were significantly associated with a shorter symptom interval (p = 0.004). A symptom interval shorter than the median symptom interval for all patients was associated with initial presentation with nausea and/or vomiting (p = 0.003), abnormal gait (p = 0.001), coordination difficulties (p = 0.006), focal motor weakness (p = 0.002), unequal pupils (p = 0.002), facial weakness (p = 0.03), and apnoea (p = 0.036), and, when grouped into combined categories, with initial presentation with any motor sign (p = 0.001). A symptom interval longer than the median symptom interval was associated with initial presentation with head tilt (p = 0.006) and cranial nerve palsies (p = 0.025). For signs and symptoms with an unknown date of onset (i.e. those other than initial ones) endocrine and growth abnormalities (p = 0.018) and reduced visual acuity (p = 0.028) were associated with a longer symptom interval Table 4.
Referral pathways
Referral pathway data were available for 101 children. Of these, 81 had visited their general practitioner, 79 a hospital paediatrician, 24 an ophthalmologist, 15 an optician and 29 had attended an emergency department. Other disciplines consulted included health visitors, orthopaedics, ear, nose and throat and speech therapy. Calculation of the number of attendances to healthcare was difficult, as records frequently did not contain details of repeated attendances to primary care. However, the reported number of attendances prior to diagnosis ranged from 0 to 12 (median 3.0) and the diagnosis of a tumour was not made in symptomatic children in both primary and secondary care. A longer than average symptom interval was significantly associated with an increased number of healthcare attendances (p < 0.001).
Discussion
This study has demonstrated a large increase in the number of presenting features between the onset of a clinical problem and diagnosis in a contemporary cohort of children with a brain tumour. Cranial nerve deficits, head tilt, endocrine and visual problems were associated with a longer symptom interval. The emergence of abnormalities of either the visual system, the motor system or of behaviour (usually lethargy) between disease onset and diagnosis was very common, suggesting the need to prioritise their reassessment in children with non-specific signs and symptoms that might be due to a brain tumour.
The median symptom interval in this cohort (3.3 months) is similar to other UK studies from the past decade but longer than that reported for large cohorts from North America [10, 14, 20]. Visual acuity is difficult to assess (and therefore may not be undertaken) in young children while identification of growth and pubertal status requires their assessment and interpretation, frequently omitted when children present to healthcare. Lethargy was the most common behavioural abnormality observed among the 56 children that had a behavioural abnormality by diagnosis and the only one present at symptom onset. There is a tendency to regard lethargy as a non-specific marker of systemic illness, however this and previous reports suggest that more emphasis should be placed on it as a specific marker of neurological illness [23]. Similarly, whilst weight loss is not a specific marker for brain tumours, just under a fifth of children had lost weight by diagnosis. Other studies have highlighted the weight loss that occurs in children with brain tumours, and the diagnostic delay that may occur whilst possible nutritional and gastrointestinal causes are investigated [16].
Although the majority of children were reviewed in primary care and general paediatrics prior to diagnosis, seven other disciplines were consulted regarding children in this cohort, highlighting the need for all healthcare practitioners to have knowledge and a high index of suspicion of brain tumour presentations. The association between symptom interval and healthcare attendances confirms that children with brain tumours present repeatedly to healthcare. Whilst children with a prolonged symptom interval will have more time to present to heath care, this suggests that diagnostic delay results from a failure to recognise signs and symptoms as being indicative of a tumour rather than a failure to seek healthcare advice.
A prolonged symptom interval is associated with an increased risk of life-threatening and disabling neurological complications at presentation and a worse cognitive outcome in survivors [3, 4, 6, 21, 26, 29]. It has a detrimental effect upon professional relationships with families and the subsequent psychological well-being of the child and their family [7]. A period of diagnostic uncertainty often precedes the diagnosis of a brain tumour, which patients and their families find extremely distressing. On being given the diagnosis, many parents report that they believe that the severity of their child's symptoms had been previously unrecognised by healthcare professionals and that pressure on their part had been necessary to make the diagnosis [7]. Parental perception that the medical response has been inadequate, incompetent or delayed may be associated with legal dispute [7]. The association between symptom interval and mortality is less clear because more biologically aggressive tumours tend to have shorter symptom intervals [3, 5, 6, 11, 13, 15].
This was a multicentre study with a short recruitment period. The recruited patients showed a similar tumour epidemiology to that reported in population registries [17, 22] and therefore the cohort reported here is likely to be representative of the current UK population of children with brain tumours. The age range and diagnoses of the 43 patients who declined participation were similar to those who were recruited, and non-participation is unlikely to have led to substantial bias. Study of signs and symptoms at initial presentation in children with tumours is only feasible in retrospect (due to the small numbers of children affected) and therefore will always be reliant on the history provided by the patient and his/her carers as recorded in the medical records at diagnosis. It is not possible to distinguish between signs and symptoms at symptom onset that were not present and those that were present but not detected. This limitation does not alter the conclusion, to which the present study leads, that those who detect the initial symptoms, whether family members, primary care or specialist health providers, should seek appropriate clinical assessment and also review after a period of time, so that additional signs and symptoms are not missed. This study is the first to report on the temporal evolution of clinical features of childhood brain tumours and suggests that the clinical problems most likely to remain unrecognised for a relatively long period are those affecting vision, other cranial nerve functions including head tilt and growth, puberty and other hormonal functions.
There are many published studies describing signs and symptoms present at diagnosis both in unselected cohorts of children with brain tumours and for specific tumour types [1, 2, 8, 9, 18, 28]. This study is the first to examine the pattern of evolution of clinical features following their onset in order to differentiate patterns of presentation requiring early referral for central nervous system imaging from those of lesser significance.
At symptom onset it may be difficult to distinguish between children with a brain tumour and those with a self-limiting benign condition, particularly as the most common initial symptoms, headache and nausea and vomiting, are known to be poor discriminators for brain tumours. This study does not provide information regarding the incidence of these clinical features in children unaffected by a brain tumour. It can therefore only identify features that are sensitive to the presence of a brain tumour, and cannot provide estimates of their specificity.
Conclusion
Children presenting with signs and symptoms that may result from a brain tumour should undergo motor and visual assessment, pubertal staging and plotting of height and weight over time against age-appropriate norms. Careful monitoring of children with abnormalities in these parameters, cranial nerve palsies and head tilt is advised. For children in whom a brain tumour is thought unlikely, the development of additional clinical features or repeated presentation should lead to a careful consideration of imaging especially if associated with abnormal visual or motor function, growth failure, focal seizures or symptoms of raised intracranial pressure.
References
Akyuz C, Emir S, Akalan N, Soylemezoglu F, Kutluk T, Buyukpamukcu M (2000) Intracranial ependymomas in childhood—a retrospective review of sixty-two children. Acta Oncol 39(1):97–100
Alston R, Newton R, Kelsey A, Newbould M, Birch J, Lawson B et al (2003) Childhood medulloblastoma in northwest England 1954 to 1997: incidence and survival. Dev Med Child Neurol 45(5):308–314
Berger C, Thiesse P, Lellouch-Tubiana A, Kalifa C, Pierre-Kahn A, Bouffet E (1998) Choroid plexus carcinomas in childhood: clinical features and prognostic factors. Neurosurgery 42(3):470–475
Chou S, Digre K (1999) Neuro-ophthalmic complications of raised intracranial pressure, hydrocephalus, and shunt malfunction. Neurosurg Clin N Am 10(4):587–608
Cohen M, Duffner P, Heffner R, Lacey D, Brecher M (1998) Prognostic factors in brainstem gliomas. Neurology 36:602–605
Comi A, Backstrom J, Burger P, Duffner P, Pediatric Oncology Group (1998) Clinical and neuroradiologic findings in infants with intracranial ependymomas. Pediatr Neurol 18(1):23–29
Dixon-Woods M, Findlay M, Young B, Cox H, Heney D (2001) Parents' accounts of obtaining a diagnosis of childhood cancer. Lancet 357:670–674
Dobrovoljac M, Hengartner H, Boltshauser E, Grotzer M (2002) Delay in the diagnosis of paediatric brain tumours. Eur J Pediatr 161(12):663–667
Dörner L, Fritsch M, Stark A, Mehdorn H (2007) Posterior fossa tumors in children: how long does it take to establish the diagnosis? Child Nerv Syst 23(8):887–890
Edgeworth J, Bullock P, Bailey A, Gallagher A, Crouchman M (1996) Why are brain tumours still being missed? Arch Dis Child 74(2):148–151
Halperin E, Watson D, George S (2001) Duration of symptoms prior to diagnosis is related inversely to presenting disease stage in children with medulloblastoma. Cancer 91(8):1444–50
http://www.statistics.gov.uk/StatBase/Product.asp?vlnk=14068&Pos=&ColRank=1&Rank=256
Kaplan A, Albright A, Zimmerman R, Rorke L, Li H, Boyett J et al (1996) Brainstem gliomas in children. A Children's Cancer Group review of 119 cases. Pediatr Neurosurg 24(4):185–192
Klein-Geltink J, Pogany L, Barr R, Greenberg M, Mery L (2005) Waiting times for cancer care in Canadian children: impact of distance, clinical, and demographic factors. Pediatr Blood Cancer 44(4):318–327
Kukal K, Dobrovoljac M, Boltshauser E, Ammann R, Grotzer M (2009) Does diagnostic delay result in decreased survival in paediatric brain tumours? Eur J Pediatr 168(3):303–310
Lehman R, Krishnamurthy S, Berlin C (2002) Weight and height deficits in children with brain stem tumours. Clin Pediatr 41:315–321
Linet M, Ries L, Smith M, Tarone R, Devesa S (1999) Cancer surveillance series: recent trends in childhood cancer incidence and mortality in the United States. J Natl Cancer Inst 91(12):1051–1058
Mehta V, Chapman A, McNeely P, Walling S, Howes W (2002) Latency between symptom onset and diagnosis of pediatric brain tumors: an Eastern Canadian geographic study. Neurosurgery 51(2):365–372
Office for National Statistics. Deaths by age, sex and underlying cause, 2003 registrations. Health Stat Q: quarterly 22
Pollock B, Krischer J, Vietti T (1991) Interval between symptom onset and diagnosis of pediatric solid tumors. J Pediatr 119(5):725–732
Reimers T, Ehrenfels S, Mortensen E, Schmiegelow M, Sonderkaer S, Carstensen H et al (2003) Cognitive deficits in long-term survivors of childhood brain tumors: identification of predictive factors. Med Pediatr Oncol 40(1):26–34
Rickert C, Paulus W (2001) Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Child Nerv Syst 17(9):503–511
Shemie S, Jay V, Rutka J, Armstrong D (1997) Acute obstructive hydrocephalus and sudden death in children. Ann Emerg Med 29(4):524–528
Stiller C (2002) Epidemiology of cancer in adolescents. Med Pediatr Oncol 39(3):149–155
Stiller C, Quinn M, Rowan S (2004) Chapter 13: childhood cancer. In: Stiller C, Quinn M, Rowan S (eds) The health of children and young people. Office for National Statistics, London
Suharwardy J, Elston J (1997) The clinical presentation of children with tumours affecting the anterior visual pathways. Eye 11(6):838–844
Wilne S, Collier J, Kennedy C, Koller K, Grundy R, Walker D (2007) Presentation of childhood CNS tumours: a systematic review and meta-analysis. Lancet Oncol 8(8):685–695
Wilne S, Ferris R, Nathwani A, Kennedy C (2006) The presenting features of brain tumours: a review of 200 cases. Arch Dis Child 91(6):502–506
Yule S, Hide T, Cranney M, Simpson E, Barrett A (2001) Low grade astrocytomas in the West of Scotland 1987–96: treatment, outcome, and cognitive functioning. Arch Dis Child 84(1):61–64
Competing interests
All authors declare that they have no competing interests.
Funding
Dr Sophie Wilne was funded by a research grant from The Big Lottery Fund in conjunction with The Samantha Dickson Brain Tumour Research Trust. The funders had no role in study design, data collection, data analysis, data interpretation or writing of this report.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wilne, S., Collier, J., Kennedy, C. et al. Progression from first symptom to diagnosis in childhood brain tumours. Eur J Pediatr 171, 87–93 (2012). https://doi.org/10.1007/s00431-011-1485-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-011-1485-7