
Abstract
Children with Down syndrome (DS) are at greater risk of pulmonary arterial hypertension (PAH) than the general population, partly due to upper airway obstruction and congenital heart disease. We wished to review our management of PAH and suggest a protocol for the systematic management of these children. Children with DS and PAH were included as referred for assessment from March 2005 to May 2010. Twenty-five patients (13 boys) met inclusion criteria. The median age was 385 days (range, 106 to 5,734); mean tricuspid regurgitation jet was 3.5 (range, 2.7–4.8) m/s. At cardiac catheterisation, mean pulmonary artery mean pressure was 26 mmHg (range, 12 to 46), and mean pulmonary vascular resistance (PVR) was 4.14 U.m2 (range, 1.20 to 12.43) at baseline. PVR fell to a mean of 2.68 U.m2 (range, 0.38 to10.69) with 20 ppm inhaled nitric oxide and 100% oxygen. Respiratory assessment included polysomnography (18), bronchoscopy (16), showing malacia (eight), adenotonsillar hypertrophy (eight) and floppy aryepiglottic folds (four). One lung biopsy showed plexogenic arteriopathy, and one was diagnosed with tracheo-oesophageal fistula. Conclusion: In order to manage this complex group of patients, a combined cardiological, respiratory and surgical approach was required. A protocol with cardiac catheterisation, blood tests and respiratory assessment is suggested for the management of pulmonary hypertension in these children.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Background
Down syndrome (DS) is the most commonly occurring genetic syndrome, and congenital heart disease (CHD) is present in approximately 50% of children with DS [6]. Atrioventricular septal defect (AVSD) is present in approximately 45% with ventricular septal defect (VSD, 35%), secundum atrial septal defect (ASD, 8%), persistent ductus arteriosus (PDA, 7%) and Tetralogy of Fallot (4%), as well as a variety of mixed lesions [15]. Some children with DS may develop further cardiovascular complications in adolescence, including mitral valve prolapse (46%) and aortic regurgitation (17%) [7].
Pulmonary arterial hypertension (PAH) can be characterised as an elevated pulmonary arterial pressure (PA pressure) and is defined as a mean PA pressure greater than 25 mmHg at rest [1] or as a tricuspid regurgitation (TR) jet greater than 2.7 m/s at echocardiography in the absence of right ventricular outflow tract obstruction [18]. Children with DS have an increased risk of developing PAH compared with the general population [8]. Exposure to increased pulmonary blood flow due to left-to-right intracardiac shunting results in increased shear stress on pulmonary endothelial cells and may impair production of nitric oxide (NO) [1]. The pathological changes are characterised by a process of vascular remodelling, eventually leading to the development of plexiform lesions and irreversible pulmonary vascular disease [14].
Amongst the numerous causes of PAH (Table 1), left-to-right shunt and chronic upper airway obstruction [16] are frequently encountered in children with DS.
Obstructive sleep apnoea [2] is common in children with Down syndrome, affecting 30–50% of this patient group compared with 3% of the general paediatric population. This may result from a number of factors, including hypotonic upper airway muscles, adeno-tonsillar hypertrophy, macroglossia, glossoptosis, flattened mid-face and narrowed nasopharynx in these patients [2]. Patients with DS are also known to be susceptible to malacic airways, in particular, laryngomalacia [5, 13]. There is increased incidence of gastro-oesophageal reflux, which may worsen airway disease [11]. Upper airway obstruction with repeated episodes of systemic hypoxaemia is associated with the development of increased pulmonary vascular resistance and may contribute to the more rapid development of PAH in children with DS and left-to-right shunts, leading to irreversible pulmonary vascular disease. The association of CHD in DS with PAH has led to neonatal screening and early intervention for cardiac lesions in DS. At the current time, it is clear that all those with large left-to-right shunts have the potential to develop pulmonary vascular disease, but some do not demonstrate any symptoms due to the raised pulmonary vascular resistance, perhaps associated with Upper Airway Obstruction (UAO) [3, 9]. This is ameliorated but not abolished by the screening programme and the advent of earlier definitive cardiac surgery at 3–6 months of age [12]. However, even with screening, a number will develop PAH and require investigation—part of the assessment of this should be a vigorous search for other causes (especially respiratory). However, despite recognition of the high prevalence for respiratory disorders in DS and their association with PAH, there have been few research reports of this specific problem. We aimed to review our experience of children with DS and PAH with a view to devising a systematic management protocol that would include appropriate investigation and management of the role of airway and respiratory disorders. It would be hoped that our attention to this subject might allow us to intervene at an early stage and modify the expected outcome in such patients.
Methods
The study was carried out at a regional tertiary referral cardiac centre in a university-affiliated children's hospital, which is the regional centre serving a population of seven million people.
Inclusion criteria
All children who had Down syndrome and pulmonary arterial hypertension confirmed on echocardiography by a tricuspid regurgitation jet > 2.7 m/s, who were referred for diagnosis and assessment between March 2005 and May 2009, were included in the study. The children were not accepted for cardiac surgical correction despite the presence of unrestrictive left-to-right shunts, since they did not display signs of cardiac failure, namely, cardiomegaly, pulmonary oedema, failure to thrive and dilated left heart chambers. They were prospectively collected on a database on the hospital computer system and confirmed by reference to Heartsuite [TM] database. This allowed for complete and accurate inclusion of patients. All parents verbally consented for inclusion of their child in this record of our experience.
Exclusion criteria
Those patients with large left-to-right shunt and high pulmonary blood flow with a low pulmonary vascular resistance on clinical and echocardiographic assessment were not referred for assessment and excluded from the study, as they would clearly benefit from surgical correction of their cardiac lesion. These children did not have cardiomegaly or pulmonary oedema on chest radiography, did not have dilated left heart chambers and were not failing to thrive. Hence, there was clinical concern that they might have high pulmonary vascular resistance, implying that cardiac operation would not be beneficial. Also, we excluded older children with established Eisenmenger syndrome in whom the diagnosis was not in doubt.
Protocol
All patients had ECG, chest radiography and cardiac catheterisation. Other echocardiographic measures were assessed in order to determine the presence and severity of pulmonary hypertension, including the degree of right ventricular dilatation, left ventricular compression, right atrial and inferior vena cava dilatation, and pulmonary regurgitation.
Respiratory investigations were determined by the respiratory physicians based on clinical judgement and included sleep study (polysomnography). If this suggested UAO, then bronchoscopy was performed at the time of cardiac catheterisation. If there were abnormalities on chest radiography, then chest CT and lung biopsy were performed as appropriate. Bronchoscopy was undertaken to exclude upper airway obstruction or to determine its cause if evidence of airway obstruction was found on polysomnography. CT scan was performed if there was evidence of interstitial lung disease, or to view the distal bronchi. Lung biopsy was performed if there was evidence of disease on the CT scan without obvious cause. Routine evaluation of all patients to exclude other causes of PAH was performed by testing for pro-coagulation abnormalities (anti-thrombin III, proteins C and S), microbiological screen (hepatitis B, C, D and E; MRSA; HIV and viral serology), immunology (including autoimmune profile, complement and anticardiolipin antibodies), and sweat test and genetic screen for BMPR2 mutations.
Cardiac catheterisation was carried out according to a standard protocol under general anaesthesia with paralysis, endotracheal intubation and femoral arterial and venous access. At baseline, an initial arterial gas was taken to ensure that the arterial carbon dioxide measurement was within normal limits. Then, measurements were made of systolic, diastolic and mean pulmonary artery pressure, mean left atrial pressure or pulmonary capillary wedge pressure and arterial pressure. Oxygen saturations (SaO2) and partial pressure of oxygen (PaO2) were recorded in the superior caval vein, inferior caval vein, pulmonary artery, left atrium (if entered) and aorta. Oxygen consumption (VO2) was also recorded. In all children, these measurements were repeated under three different conditions, with administration of 10 parts per million (ppm) NO, 20, and 20 ppm NO and 100% oxygen. From these measurements, we were able to calculate PVR, systemic vascular resistance, and the ratios.
Analysis
Results are expressed as median or mean (if normally distributed) values of the population. A paired t test was used to compare the means, and a p value <0.05 was taken as statistically significant.
Results
We identified 25 patients (13 boys) that met the inclusion criteria. It was not possible to determine the number of those who were not referred. There were of course many other children with Down syndrome and CHD who were not referred for pulmonary hypertension assessment during this period. They underwent routine surgical intervention in the usual manner, and the usual incidence of DS with CHD would apply.
All patients referred for pulmonary hypertension investigations had routine cardiac assessment in the neonatal period resulting in the following diagnoses: VSD in 11 (all unrestrictive defects with Doppler-derived pressure drop of less than 30 mmHg), complete AVSD with unrestrictive ventricular components in six and ASD in eight (ranging from 4 to 19 mm); four patients also had PDA (two restrictive and two large and unrestrictive). The median (range) age for initial assessment of PAH was 385 days (12.5 months; range, 106–5,734 days, 3 months to 15.9 years). In essence, all these cardiac conditions should have caused heart failure and high pulmonary blood flow, but did not. In many, we found that the reason for this was due to co-existing respiratory disease, as below.
Investigations
All patients had ECG changes compatible with their known cardiac pathology. Chest radiography did not demonstrate pulmonary oligaemia, pruning of peripheral vessels or cardiomegaly in any of the children. At echocardiography, all patients had a TR jet > 2.7 m/s; the mean value was 3.5 m/s (range, 2.7–4.8 m/s).
Cardiac catheterisation demonstrated a mean systolic PA pressure of 42.5 mmHg (range, 20–65), diastolic PA pressure of 13.6 mmHg (range, 5–34) and mean PA pressure of 27.5 mmHg (range, 12–46). Normal PA pressure was detected in 12 out of 25 patients during cardiac catheterisation, having been high by tricuspid regurgitation assessment on echocardiography.
The mean PVR at baseline was 4.66 U.m2 (range, 1.20–12.43). With administration of 10 ppm NO, it fell to 4.09 U.m2 (range, 0.94–13.63), and with 20 ppm NO, there was no further change (4.14 U.m2; range, 1.01–12.95). Following administration of 100% oxygen, the PVR fell to 2.99 U.m2 (range, 0.38–10.69). The ratio of pulmonary to systemic flow at baseline (Q p/Q s) had a mean value of 1.49 (range, 0.53–4.21).
Twelve of the 18 patients who underwent PSG had evidence of UAO. Sixteen underwent bronchoscopy, revealing tracheo- or broncho-malacia in eight, floppy aryoepiglottic folds in four and large tonsils and adenoids in another eight. One child had previously undiagnosed trachea-oesophageal fistula requiring surgical repair. CT scan, performed in four patients showed interstitial disease in one patient, and lung biopsy revealed arteriopathic changes of uncertain cause.
Management—cardiac
Those 18 patients whose PVR was <7 U.m2 and where it fell by more than >20% in response to NO underwent intervention with cardiac surgery or cardiac catheterisation, all of whom survived.
Management—airways
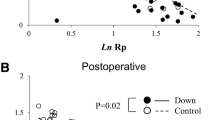
Eighteen out of the 25 patients had airways disease; of these, 12 had normal pulmonary artery pressures at the time of cardiac catheterisation. Those patients with airway disease had a lower PA pressure at cardiac catheterisation than estimated from echocardiography (Fig. 1). The mean PA pressure at baseline for the patients in the group with airways disease was 23 mmHg (range, 17–30 mmHg), compared with a value of 34 mmHg (range, 17–39) in the patients without airway obstruction (p < 0.05).

Comparison of PA pressures in response to oxygen and nitric oxide in patients with and without airways obstruction
In 12 out of the 25 patients (14 out of the 30 catheterisation procedures), aortic oxygen saturations decreased with administration of 10 ppm NO.
Eight patients were found to have clinically significant laryngo-tracheo-bronchomalacia; five were managed using continuous positive airways pressure (CPAP), and four have had an aryepiglottoplasty. One patient was found to have a tracheoesophageal fistula, which was repaired surgically. Eight patients had adeno-tonsillar hypertrophy, all of whom have had a fall in their pulmonary artery pressure after surgical intervention. One patient was found to have plexogenic arteriopathy on lung biopsy.
Drugs
In those with evidence of night-time hypoxia from sleep study, overnight oxygen therapy was used (12 patients). The treatment time ranged from 6 months to 2.1 years. Although the data is limited on the effectiveness of disease-modifying therapy in those with airways or respiratory disease, 12 were managed with sildenafil at a dose of 0.5–1 mg/kg 8-hourly.
Outcome
The mean length of follow-up in this study was 3.9 years (range, 0.85–5.2). All patients continue to receive routine cardiac follow-up. Respiratory symptoms resolved in eight out of the 12 patients with initial respiratory problems and were persistent in four. One of these patients has irreversible PVD and recurrent chest infections and the other three have airway malacia. These patients continue to be monitored on an outpatient basis.
Pulmonary hypertension improved in 21 out of 25 patients, demonstrated as a reduction in the TR jet between baseline and follow-up (Fig. 2). The TR jet remained constant in three patients and increased in one. Five patients underwent repeat cardiac catheterisation (of whom one patient had two further catheterisation procedures); mean PA pressure decreased in two, remained constant in one and increased in one patient.

Change in TR jet between baseline and follow-up
Eleven out of 25 patients had a TR jet ≤ 2.7 m/s at follow-up (p = 0.0007, compared with the values at entry), representing a resolution of their pulmonary hypertension. The child whose TR jet value increased during the study period is an interesting case of a girl who had DS, PAH, a VSD and upper airway disease. Her PAH improved with treatment, but she then developed a severe form of arthritis, and this appears to have been associated with a worsening of her PAH.
Discussion
We have shown that when children with Down syndrome, congenital heart disease and airway disease are managed by a multidisciplinary team, a high prevalence of associated respiratory problems is identified. Many of those children, who, on clinical grounds, did not have signs of a large left-to-right shunt, were found to be entirely suitable for cardiac surgical correction, once a cardiac catheterisation had been performed under general anaesthetic and their airway problem identified. It is important that children with Down syndrome are screened for congenital heart disease, regardless of symptoms. In this population, these investigations have allowed us to diagnose a number of airway diseases in these children in a systematic manner. Our group of 25 children had a variety of cardiac and respiratory complications which have been successfully managed with good outcome.
Although it is well recognised that there may be cardiac and respiratory disease with pulmonary hypertension in children with Down syndrome, there is little information in the literature on how these should be approached together. The diagnosis of PAH in children with Down syndrome is now important, since there are a number of disease-targeted therapies available, including intravenous epoprostenol (the only drug for children tested in a placebo-controlled trial), bosentan and sildenafil [19]. These therapies may be used pre-operatively, post-operatively or palliatively. Currently, there are no national guidelines regarding the medical management of patients with PAH and DS. In practice, many of the methods in use in children without DS are often employed. Ultimately, some patients may need to undergo transplantation, although referral rates remain low amongst patients with DS, in part, due to concerns regarding post-operative complications and post-transplant malignancy secondary to immuno-suppression [10].
In four children, an aryepiglottoplasty was performed; in one, we found arteriopathic disease, and in eight, adeno-tonsillectomy was performed. Eleven children underwent treatment with home oxygen. During the period of study, pulmonary hypertension resolved in 11 out of the 25 patients, fell in nine, did not change in three and increased in the one patient who developed severe arthritis. In total, 12 underwent intervention to manage their airway problems.
Echocardiography demonstrated that all children in this study had pulmonary hypertension, which is how they were selected, whereas at cardiac catheterisation, 12 children, who were found to have abnormal PSG, had normal pulmonary artery pressures. One possible explanation is that placement of an endotracheal tube relieved the upper airway obstruction and temporarily resolved the pulmonary hypertension, which would account for the predominance of confirmed airway obstruction in eight of these patients.
Our study also suggests that increasing the dose of NO from 10 to 20 ppm has little additional effect. The maximal vasodilatory response in these patients can be achieved by addition of oxygen. This might save time and potentially dangerous additional exposure to anaesthesia for these patients.
Interestingly, our results have suggested that in some patients, administration of NO leads to a decrease in systemic oxygen saturation. One possible explanation might be that administration of NO causes vasodilatation of the pulmonary vasculature and greater pulmonary blood flow without greater oxygen uptake and hence, lower oxygen saturation. The reason why this takes place in some patients and not in others remains unclear. It may be that those patients who demonstrate a fall in oxygen saturations in response to NO may have concurrent airway disease contributing to their PAH, explaining why vasodilatation alone does not cause an increase in oxygenation. This hypothesis is supported by the high prevalence of airway disease in this subset of patients. Alternatively, the vasodilatory effects of NO may simply be more apparent in those patients with a greater degree of pulmonary vasoconstriction at baseline (effectively, those with more significant pulmonary vascular disease). This may suggest a role for the use of NO administration as a screening tool to determine the extent to which airways disease is contributing to pulmonary hypertension, but further studies are needed.
Currently, there is limited research regarding pulmonary hypertension in Down syndrome and little advice on how this condition should be managed. It is recommended that, to ameliorate the symptoms of airway disease and to prevent the development of PAH, patients with DS should have aggressive treatment of cardiac disease, gastro-oesophageal reflux disease and upper airway disease [4].
Current best practice suggests that prompt surgical correction of cardiac defects is imperative to prevent the development of irreversible pulmonary vascular disease [12]. The mainstay of management of lower airways disease is the use of prophylactic antibiotics and regular inhaled corticosteroids, alongside oxygen and physiotherapy [4]. Adenotonsillectomy may be beneficial in those patients with significant airways obstruction [4], and children with severe laryngo-tracheo-bronchomalacia may require treatment with home oxygen therapy, with or without CPAP [4], or even aryepiglottoplasty [11].
A strength of the study is that the data was collected on an individual database, by one operator. Hence, there is complete data acquisition, with all the patients undergoing the same cardiac catheterisation testing protocol. However, the number of patients is not large and will be increased with greater experience. Also, we cannot be certain that these children would not have improved with time, without any airways intervention. In order to approach this, a multi-centre randomised trial would be necessary. However, we know that the chronic hypoxia resulting from upper airways obstruction in children with Down syndrome contributes to the development of pulmonary hypertension [2]. It may therefore be unethical not to treat pulmonary hypertension in these patients, and a trial would have to ensure the maximum safety of the patients under current knowledge.
In light of our findings and the evidence currently available, we suggest an investigation and management protocol for children with Down syndrome and pulmonary hypertension (Fig. 3).

Suggested management protocol for patients with DS
Notably, the high prevalence of asymptomatic obstructive airway disease in children with DS and the risks of developing irreversible PAH support routine sleep studies in children with DS, even those without symptomatic airway disease [17].
The relative contributions of cardiac and airways disease in children with DS and pulmonary hypertension have yet to be determined. We have also yet to establish the mechanism responsible for the fall in oxygen saturations seen in some patients with airways disease and administration of nitric oxide. Crucially, further studies are required to facilitate the development of guidelines to establish which drugs are most appropriate for the management of PAH in this group of children. We suggest that children with PAH and DS are assessed and managed by a multidisciplinary approach combining cardiological and respiratory assessment and present a suggested protocol for further evaluation. However, it is still not entirely clear whether a more aggressive cardiac or respiratory approach to these children is correct. A randomised prospective multi-centre study would need to be performed to ascertain the contribution of the different cardiac morphologies and respiratory diseases to this group of children.
References
Andrews R, Tulloh R (2002) Pulmonary hypertension in pediatrics. Curr Opin Pediatr 14:603–605
de Miguel-Diez J, Villa-Asensi J, Alvarez-Sala JL (2003) Prevalence of sleep-disordered breathing in children with Down syndrome: polygraphic findings in 108 children. Sleep 26:1006–1009
Down’s Syndrome Medical Interest Group (DSMIG) (2007) Basic medical surveillance essentials for people with Down’s syndrome—cardiac disease: congenital and acquired. Available at http://www.dsmig.org.uk/. Accessed February 2009
Down’s Syndrome Medical Interest Group (DSMIG) (2001) Respiratory disorders with Down’s syndrome: overview with diagnostic and treatment options. 2001. Available at http://www.dsmig.org.uk/. Accessed February 2009
Eipe N, Lai L, Doherty D (2009) Severe pulmonary hypertension and adenotonsillectomy in a child with Trisomy-21 and obstructive sleep apnoea. Ped Anesth 19:541–553
Freeman SB, Taft LF, Dooley KJ et al (1998) Population-based study of congenital heart defects in Down syndrome. Am J Med Genet 80:213–217
Geggel RL, O’Brien JE, Feingold M (1993) Development of valve dysfunction in adolescents and young adults with Down syndrome and no known congenital heart disease. J Pediatr 122:821–823
Greenwood RD, Nadas AS (1976) The clinical course of cardiac disease in Down’s syndrome. Pediatrics 58:893–897
Kawai T, Wada Y, Enmoto T et al (1995) Comparison of hemodynamic data before and after corrective surgery for Down’s syndrome and ventricular septal defect. Heart Vessels 10:154–157
Leonard H, Eastham K, Dark J (2000) Heart and heart lung transplantation in Down’s syndrome. BMJ 320:816–817
Martin JE, Howarth KE, Khodaei I et al (2005) Aryepiglottoplasty for laryngomalacia: the Alder Hey experience. J Laryngol Otol 119:958–960
Masuda M, Kado H, Tanoue Y et al (2005) Does Down syndrome affect the long-term results of complete atrioventricular septal defect when the defect is repaired during the first year of life? Eur J Cardiothorac Surg 27:405–409
Mitchell RB, Call E, Kelly J (2003) Diagnosis and therapy for airway obstruction in children with Down syndrome. Arch Otolaryngol Head Neck Surg 129:642–645
Rabinovitch M, Keane JF, Norwood WI et al (1984) Vascular structure in lung tissue obtained at biopsy correlated with pulmonary hemodynamic findings after repair of congenital heart defects. Circulation 69:655–667
Roizen NJ, Patterson D (2003) Down’s syndrome. Lancet 361:1281–1289
Rowland TW, Nordstrom LG, Bean MS et al (1981) Chronic upper airway obstruction and pulmonary hypertension in Down’s syndrome. Am J Dis Child 135:1050–1052
Shott SR, Amin R, Chini B et al (2006) Obstructive sleep apnea: should all children with Down syndrome be tested? Arch Otolaryngol Head Neck Surg 132:432–436
Tulloh R (2005) Congenital heart disease in relation to pulmonary hypertension in paediatric practice. Paediatr Respir Rev 6:174–180
Tulloh R (2009) Etiology, diagnosis and pharmacologic treatment of pediatric pulmonary hypertension. Paediatr Drugs 11:115–128
Conflict of interest
This study was not funded, and there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hawkins, A., Langton-Hewer, S., Henderson, J. et al. Management of pulmonary hypertension in Down syndrome. Eur J Pediatr 170, 915–921 (2011). https://doi.org/10.1007/s00431-010-1378-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-010-1378-1