
Abstract
Hyper IgE syndrome (HIES) is a rare primary immunodeficiency disorder, characterized by eczema, recurrent skin and lung infections, and significantly elevated serum IgE level. It was previously diagnosed based on clinical manifestations and laboratory markers that were not specific to the disease. Recent studies have demonstrated that mutations in signal transducer and activator of transcription 3 (STAT3) cause the autosomal dominant or sporadic HIES, which make the disease definitively characterized at molecular level. Here, we reported a 3-year old Chinese boy with neonatal-onset rash and multiple serious Staphylococcus aureus infections including recurrent skin abscesses, liver abscess, sepsis, and destructive pulmonary infection (pneumonia, multiple pulmonary abscesses, pyopneumothorax, and finally, pneumatocele). Genetic study revealed a heterozygous mutation in exon 21 of STAT3 gene (g.66583 A > C, c.1970A > C) in the boy, which resulted in a substitution of tyrosine at the amino acid position 657 to serine (p.Y657S) in the Src homology 2 (SH2) domain of STAT3. Functional prediction with bioinformatics programs of the Sorting Intolerant from Tolerant (SIFT) and the Polymorphism Phenotyping (PolyPhen) reported “deleterious (SIFT score 0.02)” and “probably damaging (PSIC score difference 2.94)” values, respectively. Further study of family members revealed that neither his parents, nor his twin brother carried the mutation, indicating the mutation was likely to occur de novo in our patient. Conclusion: The mutation,p.Y657S,in SH2 domain of STAT3 is a disease-causing mutation in the boy with HIES.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hyper-IgE syndrome (HIES; OMIM 147060) is a rare primary immunodeficiency disorder characterized by eczematoid dermatitis, recurrent cutaneous and pulmonary infections, and significantly elevated serum IgE level. It was first reported as Job's syndrome by Davis et al. in 1966 [2], and further characterized by Buckley et al. in 1972 [1]. HIES can be subdivided into two distinct types: type 1 HIES, which is the classic autosomal dominant (AD-HIES) or sporadic, and type 2 HIES, which is autosomal recessive (AR-HIES). Clinically, type 1 HIES displays abnormalities in multiple systems, including skeletal/dental, connective tissue, and immune systems, whereas abnormalities of type 2-HIES are confined to the immune system [9, 13]. In 2006, Minegishi et al. identified a tyrosine kinase 2 (TYK2) deficiency in an AR-HIES patient associated with susceptibility to intracellular bacterial and viral infections [9]. Afterwards, the mutation in dedicator of cytokinesis 8 (DOCK8) was demonstrated to be responsible for many cases of AR-HIES [3, 21]. In the year 2007, Holland et al. [6] and Minegishi et al. [10] independently identified that mutations in the gene, encoding the signal transducer and activator of transcription 3 (STAT3), are associated with type 1 HIES. Here, we reported a Chinese boy presented with neonatal-onset eczematoid rash and recurrent serious staphylococcal infections. A novel mutation in the STAT3 gene was identified in him.
Case report
A three-year-old boy from rural area was admitted due to fever, cough, and tachypnea for 10 days. He had suffered from refractory eczematoid dermatitis since he was a newborn and was hospitalized once, due to complicated skin infections. He also had recurrent infections, including 18 upper respiratory infections and multiple episodes of oral candidiasis. At the age of 18 months, he presented with fever and abdominal pain and was diagnosed with a staphylococcal liver abscess (Fig. 1A), which was treated with surgical drainage and antibiotics. Since then, he has been plagued by recurrent scalp boils and skin abscesses, and drainage incisions have been made in his neck and armpit four times. The patient and his twin brother were delivered at 36 weeks by cesarean section and were found that each have their own placenta, suggesting they were fraternal twins. The birth weight of the patient was 2,100 g. He has not accepted any vaccine inoculation due to recurrent skin and respiratory infections. His parents were not consanguineous, and there was no family history of relevant allergic or immunologic diseases. His dizygotic twin brother is healthy.

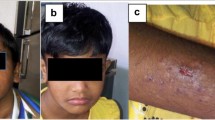
Clinical information of the patient. (A) Abdominal CT of the patient showed multiple abscesses in the right lobe of the liver. (B) The boy's photo showed the coarse face, eczema, and boils on his scalp. (C–E). Series chest CT of the patient showed multiple pulmonary abscesses, pneumopyothorax, and pneumatocele
Physical examination revealed fever, tachycardia, and tachypnea. His scalp was covered with impetigo, and he suffered from exanthema papulosum (Fig. 1B). Facial eczematous dermatitis, flesh nasal tip, and mild broad nasal bridge made a “coarse” appearance. Chest examination revealed tracheal deviation to the left, a full right thorax, bilateral asymmetry of breath movement, and the breath sound on the right had almost disappeared. Crackles in the left lung were also noted. The other clinical manifestations were included in Table 1.
Laboratory findings were as follows: WBC, 29.5 × 109/L ∼42.2 × 109/L; neutrophils, 11.3 × 109/L ∼18.2 × 109/L; eosinophils,10.2 × 109/L ∼13.50 × 109/L; lymphocytes, 2.95 × 109/L ∼6.05 × 109/L; Hb, 94 g/L ∼104 g/L; platelets, 629 × 109/L ∼747 × 109/L; CRP, 17 mg/L; Serum IgE, 883 IU/ml (0 ∼60 IU/ml); IgM, 4.38 g/L (elevated); and, IgG, IgA, and complement C3 and C4 were all normal. The nitroblue tetrazolium (NBT) test was normal. Furthermore, T lymphocyte subsets were also normal (CD4 2.39 × 109/L and CD8, 1.38 × 109/L, respectively). Two separate blood cultures tested positive for bacteria; one cultured contained Staphylococcus aureus and Pseudomonas aeruginosa, and one was positive for only S. aureus. S.aureus was also isolated from empyema drain of the patient. Series chest X-ray and CT of the patient indicated staphylococcal pneumonia, multiple pulmonary abscesses, right pyopneumothorax, and finally, pneumatocele (Fig. 1C–E) after surgical drainage and treatment with antibiotics (vancomycin and cefepime hydrochloride, intravenously). Based on these clinical and laboratory findings, the type 1-hyper-IgE syndrome was suspected. The scoring system of National Institutes of Health (NIH) for hyper-IgE syndrome was used [5], and the score of the patient is 58 points, as shown in the table.
Written informed consent was obtained from the parents for a molecular genetic study of STAT3 and the use of the boy's images. This study was also approved by the Ethic Committee of West China Second University Hospital. Peripheral blood was collected (EDTA-coagulated) from a healthy volunteer, the patient, his twin-brother, and his parents. The genomic DNA and total RNA were prepared, and all 24 exons and exon/intron boundaries in the STAT3 gene were separately amplified from the patient's genomic DNA, using PCR and specific oligonucleotide primers (available on request). RT-PCR was also performed as described previously [6]. The PCR products were directly sequenced using the ABI Big Dye Terminator mix (Applied Biosystems) and 3730XL DNA sequencer (Applied Biosystems). The results were analyzed using Sequencing Analysis Software v5.2 (Applied Biosystems). A heterozygous mutation in exon 21 (g.66583 A > C, c.1970A > C ) was identified in the boy (Fig. 2B and C), which resulted in a substitution of tyrosine at the amino acid position 657 to serine (p.Y657S) in the Src homology 2 SH2 domain of STAT3. This mutation was not found in the single nucleotide polymorphism database (dbSNP; www.ncbi.nih.gov/projects/SNP/). Further study of family members revealed that neither his parents, nor his twin brother carried the mutation, indicating the mutation of STAT3 is likely occurred de nove in our patient (Fig. 2A, D, E and F). We also observed that p.Y657S variation did not occur in 50 control chromosomes from 25 healthy individuals. Comparative genomics analysis, by aligning nucleotide sequences of different species, such as Rattus norvegicus (Norway rat), Mus musculus (house mouse), Canis lupus familiaris (dog), Pan troglodytes (chimpanzee), and Gallus gallus (red jungle fowl), showed that the Y657 in the SH2 domain of STAT3 is perfectly conserved. Functional predictions of the p.Y657S mutation were performed by use of bioinformatics methods encoded in the programs Sorting Intolerant from Tolerant (SIFT, http://blocks.fhcrc.org/sift/SIFT.html) and Polymorphism Phenotyping program (PolyPhen, http://genetics.bwh.harvard.edu/pph). The SIFT reported “deleterious (SIFT score 0.02)” and Polyphen reported “probably damaging (PSIC score difference, 2.94)” values, respectively.

STAT3 mutation analysis in the patient with hyper-IgE syndrome showed a heterozygous mutation in exon 21 of STAT3 gene (g.66583 A > C, c.1970A > C), which lead to the mutation of p.Y657S in the SH2 domain of STAT3 (B). The reverse sequencing further confirmed the mutation (C). The healthy volunteer (A), the patient's twin brother (D) and his parents (E, F) were normal
Discussion
HIES is a rare primary immunodeficiency disease with two distinct phenotypes. The clinical discriminants between two types are that type 1 HIES manifests as characteristic coarse facies, skeletal/connective tissue abnormalities (pathological fracture, retention of deciduous teeth, scoliosis, and hyperextensibility), and pneumatocele after pulmonary staphylococcal infection, while type 2 HIES presents with severe viral infections, central nervous system involvement, and intracellular bacterial infections, but absence of skeletal/connective tissue abnormalities and pneumatocele [11, 13]. Although some cases of familial type 1 HIES with autosomal dominant inheritance have been reported, most cases of type 1 HIES (> 90%) are sporadic [11]. Clinically, almost all patients with HIES suffer from recurrent staphylococcal infections, usually beginning in infancy, and predominantly, confined to the skin and lungs [11]. This situation contrasts starkly with chronic granulomatous disease (CGD), in which the NBT test is negative, and recurrent staphylococcal infection occur in a wide variety of organs [11, 18]. Our patient not only has devastating pulmonary staphylococcal infection and recurrent skin abscesses, but also liver abscess, which is rare in patients with HIES, only two cases were reported in literature [4, 15].
HIES was previously diagnosed on the basis of NIH scoring system [5], but the scoring based on clinical manifestations and laboratory markers (serum levels of IgE and eosinophilia) was not specific to the disease, especially for young children. Some clinical manifestations of HIES, such as retained primary teeth, pathologic bone fractures, and lymphoma are usually seen in elder children and adults. The discovery of mutation in STAT3 gene causing type 1 HIES, definitively characterize the disease at molecular and immunologic levels. STAT3 is a transcription factor that binds to the STAT3 responsive elements in the promoter regions of various genes. STAT3 not only plays a critical role in the signal transduction pathway of multiple cytokines, including the IL-2/γc cytokines, IL-6/gp 130 cytokines, type-1 and type-2 interferons (IFNγ,IFN-α/β), IL-10 family cytokines, and receptor tyrosine kinases, etc. [11, 17], but also has specific roles in organogenesis, organ preservation, and organ-specific inflammation [6]. Recent studies have demonstrated that STAT3 has a significant impact on development of IL-17-producing T helper cells (TH17 cells), which are critical in the clearance of fungal and extracellular bacterial infections [8]. Minegishi et al. observed that human keratinocytes and bronchial epithelial cells, unlike other cells, were more dependent on TH17 cytokines (IL-17 and IL22) for the production of antistaphylococcal factors, including the neutrophil-recruiting chemokines and antimicrobial peptides including β-defensins [12], the latter plays a crucial role in killing Staphylococci [16]. These findings may explain why systemic TH17 deficiency in HIES patients with staphylococcal infection restricted to the skin and lungs [12]. The deficiency of TH17 cells caused by STAT3 mutation could be the underlying cause of increased susceptibility to recurrent infections commonly seen in HIES. Based on molecular immunological advances, a new diagnostic guidelines for STAT3-deficient HIES (i.e., AD-HIES or sporadic cases) was proposed by Woellner et al. in 2010 [19]. Our patient can be diagnosed as STAT3-deficient HIES according to the guidelines.
To date, the STAT3 mutations reported in type 1 HIES patients are predominantly located in DNA binding domain (DBD) and SH2 domain [6, 7, 10, 14, 19]. Some “hot spots” have been reported [6, 7, 10, 14, 19] . The mutations in DBD result in a protein with an impaired ability to target DNA sequences in promoter region but can still interact with a wild-type STAT3 protein and form nonproductive, dominant-negative STAT3 dimers. The mutations affecting the SH2 domain impair STAT3 dimerization and results in a similar reduction in target-gene expression [6]. At the C-terminal end of STAT3, there is a converse residue (Y705) that can be phosphorylated (pY705), while in SH2 domain of STAT3, there is a phosphorylated tyrosine (pY) peptide-binding pocket that consists of three sites—the pY-residue binding site, the +3 residue-binding site, and a hydrophobic binding site (W623,Q635,V637,Y640,Y657) [20]. The dimerization of STAT3 occurs through reciprocal SH2-pY705 motif interactions, followed by nuclear translocation, binding to specific DNA elements, and upregulation of target genes[7,14,]. Xu et al. demonstrated that the pocket was pivotal for the function of STAT3 [20]. In this article, we report a heterozygous mutation in SH2 domain of STAT3, p.Y657S (g.66583 A > C, c.1970A > C), which might affect the structure of the hydrophobic binding site in this crucial pocket, preventing the dimerization of STAT3. Functional prediction of the mutation with the SIFT program reported “deleterious (SIFT score 0.02)”, and the PolyPhen program reported “probably damaging (PSIC score difference 2.94)”, respectively, indicating p.Y657S is with high confidence supposed to affect STAT3 protein function or structure. To our knowledge, this mutation has not yet been reported in literature, only two patients with p.Y657C (c.1970A > G ) mutation in STAT3 has been reported [6, 19] (the same patients were included in the literatures reported by Holland et al. [6] and Woellner et al. [19], respectively).Our patient is unusual in that he had multiple serious Staphylococcus aureus infections in only 3-year-old age, including recurrent skin abscesses, liver abscess, sepsis, and especially, destructive pulmonary infection, including pneumonia, pulmonary abscesses, pyopneumothorax, and finally, pneumatocele, suggesting the mutation p.Y657S in SH2 domain of STAT3 is a disease-causing mutation.
References
Buckley RH, Wray BB, Belmaker EZ (1972) Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics 49:59–70
Davis SD, Schaller J, Wedgwood RJ (1966) Job's syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet 1:1013–1015
Engelhardt KR, McGhee S, Winkler S et al (2009) Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. Allergy Clin Immunol 124:1289–1302.e4
Fanconi S, Seger RA, Willi U et al (1984) Oral chloramphenicol therapy for multiple liver abscesses in hyperimmunoglobulinemia E syndrome. Eur J Pediatr 142:292–295
Grimbacher B, Schäffer AA, Holland SM et al (1999) Genetic linkage of hyper-IgE syndrome to chrosome 4. Am J Hum Genet 65:735–744
Holland SM, DeLeo FR, Elloumi HZ et al (2007) STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 357:1608–1619
Jiao H, Tóth B, Erdos M et al (2008) Novel and recurrent STAT3 mutations in hyper-IgE syndrome patients from different ethnic groups. Mol Immunol 46:202–206
Milner JD, Brenchley JM, Laurence A et al (2008) Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452:773–776
Minegishi Y, Karasuyama H (2007) Hyperimmunoglobulin E syndrome and tyrosine kinase 2 deficiency. Curr Opin Allergy Clin Immunol 7:506–509
Minegishi Y, Saito M, Tsuchiya S et al (2007) Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448:1058–1062
Minegishi Y (2009) Hyper-IgE syndrome. Curr Opin Immunol 21:487–492
Minegishi Y, Saito M, Nagasawa M et al (2009) Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J Exp Med 206:1291–1301
Renner ED, Puck JM, Holland SM et al (2004) Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. J Pediatr 144:93–99
Renner ED, Rylaarsdam S, Anover-Sombke S et al (2008) Novel signal transducer and activator of transcription 3(STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol 122(1):181–187
Roy AD, Mcllrath EM, Mawhinney H (1982) Multiple liver abscesses in a patient with hyper IgE syndrome. J R Coll Surg Edinb 27:224–227
Sass V, Schneider T, Wilmes M et al (2010) Human beta-defensin 3 inhibits cell wall biosynthesis in Staphylococci. Infect immune 78:2793–2800
Tangye SG, Cook MC, Fulcher DA (2009) Insight into the role of STAT3 in human lymphocyte differentiation as revealed by the hyper-IgE syndrome. J Immunol 182:21–28
Winkelstein JA, Marino MC, Johnston RB Jr et al (2000) Chronic granulomatous disease. Report on a national registry of 368 patients. Med Baltim 79:155–169
Woellner C, Gertz EM, Schäffer AA et al (2010) Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome. J Allergy Clin Immunol 125:424–432.e8
Xu X, Kasembeli MM, Jiang X et al (2009) Chemical probes that competitively and selectively inhibit STAT3 activation. PLoS ONE 4:e4783
Zhang Q, Davis JC, Lamborn IT et al (2009) Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med 361:2046–2055
Acknowledgments
Qiang Li has received a research funding from the Department of Science and Technology of Sichuan Province, China (No.2008JY0029-1). We deeply thank Professor Yu-lung Lau*, Professor Wen-wei Tu* and Dr.Koon-wing Chan* for their invaluable help (Qiang Li has received a 3-months training in molecular diagnosis of primary immunodeficiency disease in the Department of Paediatrics & Adolescent Medicine, Hong Kong University). We are also grateful to the patient and his parents for their cooperation.
*Department of Paediatrics & Adolescent Medicine, Hong Kong University.
Conflict of Interest Statement
Qiang Li has received research support from the Department of Science and Technology of Sichuan Province, China (No.2008JY0029-1). All authors declare that no actual or potential conflict of interest in relation to this article exists.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, Jy., Li, Q., Chen, Tt. et al. Destructive pulmonary staphylococcal infection in a boy with hyper-IgE syndrome: a novel mutation in the signal transducer and activator of transcription 3 (STAT3) gene (p.Y657S). Eur J Pediatr 170, 661–666 (2011). https://doi.org/10.1007/s00431-010-1349-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-010-1349-6