
Abstract
Localised duplications, involving the MECP2 locus, at Xq28 have been associated with a syndrome comprising X-linked mental retardation, hypotonia and recurrent infections in males. We now present neuroradiological evidence that progressive cerebellar degenerative changes may also be a consistent feature of this syndrome, emerging in the second decade of life. We report seven affected males, from three different families who, in addition to the previously described clinical findings, have a reduction in the volume of the white matter and mild dilatation of the lateral ventricles. Three of the older patients show a consistent cerebellar degenerative phenotype. Furthermore, we describe the first female affected with the disorder. The female was mildly affected and shows X-inactivation in the ratio of 70:30, demonstrating that X-inactivation cannot be exclusively relied upon to spare the female carriers from symptoms. In conclusion, there is a radiological phenotype associated with Xq28 duplication which clearly demonstrates progressive degenerative cerebellar disease as part of the syndrome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
While in excess of 20 loci for non-syndromic X-linked mental retardation have been identified on the X chromosome, the prevalence of mutation at individual gene level appears to be quite low. Moreover, many of the affected individuals identified do not have characteristic clinical features, which would point the investigating clinician towards a syndromic locus as the root cause of diagnosis [9]. The newly emerging syndrome of Xq28 duplication syndrome represents an important likely exception to the clinically amorphous group which X-linked non-specific mental retardation represents. The emergence of the syndrome derives in part from the development of array comparative genome hybridisation of an 80-kb resolution for the X chromosome to screen patients with suspected X-linked mental retardation but whose clinical phenotypes were non-specific [11]. Using this approach, a small duplication of Xq28, encompassing L1CAM and MECP2, was identified in a large family with severe X-linked mental retardation associated with spasticity. Applying the same technique to a wider panel of 17 further cases of non-specific mental retardation, three additional cases of Xq28 duplication were recognised. The work of Frietz et al. [4] also constituted an important landmark in the emergence of this condition as a recognisable syndrome, these authors reporting six additional affected pedigrees and emphasising the clinical features common to those pedigrees of infantile hypotonia, recurrent respiratory infection, severe mental retardation, absence of speech, seizures and spasticity. A more recent study of 283 patients with X-linked mental retardation identified a further three cases (prevalence 1%), while three further examples of MECP2 duplication were established in a population of 134 male patients with mental retardation and severe, mostly progressive, neurological symptoms [7].
We now report three recently identified and previously unreported families in whom the availability of serial clinical data and neuroradiological images over several years afford novel observations which may assist clinicians in resolving previously unrecognised cases of this emerging condition.
Family 1
The structure of the family is shown (Fig. 1). Case III2 first came to paediatric attention at age 10 months when he presented with a left-sided pneumococcal infection with massive pleural effusion, which required drainage. His infection required intensive management and follow-up, during which it was noted that his development was delayed with respect to his dizygotic twin brother. Mild hypotonia was recorded at age 1 year, and hyper-reflexia of the lower limbs was also noted by a paediatric neurologist. Seizures commenced at age 4 years and took the form of stiffening with eye rolling and clonic movements. These proved refractory to control despite many different combinations of medication. Having walked at age 3 years, he had lost this facility by age 10 years and has required nasogastric feeding since age 13 years. He never acquired speech. Clinical examination at age 18 years is of a non-dysmorphic, thin boy, who shows no evidence of response to surroundings and whose lower limbs are hyper-reflexic. His parents particularly draw attention to episodes of hyper-pyrexia and shivering, unrelated to infection, and suggest that his temperature control is impaired. There have been several episodes of serious chest infections, which have required hospital admission and intensive intervention with antibiotic support, physiotherapy and oxygen supplementation.

The pedigree of family 1
His sister, III4, is also non-specifically developmentally delayed. She is non-dysmorphic, has not had seizures and has not experienced temperature dysregulation phenomena nor severe chest infections. Neurological examination, at age 12 years, betrays no abnormality of tone, good strength and well-coordinated locomotion. She speaks fluently but her intellectual performance has always been a cause for concern and she attends a special school where, with intensive input, she makes discernible progress. There has been no evidence of seizure activity.
His cousin, case III7, came to attention early with developmental delay. Pregnancy had been normal and birth weight was 3.6 kg. Developmental delay was noted early. Head control was not established until 8 months of age, and he learned to sit by 18 months of age, with intensive physiotherapy. At age 4 years, he was walking independently and later learned to climb stairs holding on but was always unable to descend stairs. Having previously fed independently with a spoon, by 8 years, he was feeding by hands only. He recognised family members, understood simple commands but had no words. He never achieved independent toileting. His course had been characterised by severe respiratory infections requiring multiple admissions from early childhood. On several occasions, he required ventilation in his local hospital for seemingly innocuous chest infections, so severe was his response to infectious challenge. Seizures were observed from age 7 years, initially presenting as drop attacks without loss of consciousness. Thereafter, this aspect progressed to involve nocturnal seizures occurring in clusters several times a night. When examined at age 8 years, occipitofrontal circumference was documented as 55 cm (97th centile) and posture was abnormal with waddling gait and lordosis. There were rapid jerky movements and tremulousness of the limbs. Tone was deemed to be normal and deep tendon reflexes were reduced. Plantar responses were flexor. Electroencephalogram showed generalised discharges with a right-sided predominance. Computed tomography (CT) scan of the thorax did not show any evidence of bronchiectasis. He developed evidence of regression from age 10 years, with progressive weakness and ceased walking from age 11. Swallowing also deteriorated at this time, and for the last year of his life, he was only able to swallow a little mashed food. He died aged 13 years and 10 months from a severe pneumonia, which proved refractory to antibiotics.
Radiological findings
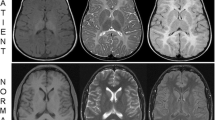
Both boys have a reduction in the volume of white matter and also increased T2 and decreased T1 signal in the deep white matter and mild dilatation of the lateral ventricles particularly at level of atria and occipital horns. The appearances mimic those of periventricular leucomalacia. These observations are in films taken when the patients were 8 and 6 years old, respectively, and the posterior fossa was normal in both cases. Further magnetic resonance imaging (MRI) studies of III2 at age 14 years show abnormal increased signal in the deep periventricular white matter, a generalised reduction in white matter volume (Fig. 2) and the emergence of progressive inferior cerebellar atrophy (Fig. 3). Creatine level is normal on spectroscopy. MRI of the symptomatic female at age 10 years was unremarkable.

Axial T2 fluid attenuated inversion recovery (FLAIR) image on case III2 (family 1), at age 17 years showing very mild dilatation of the atria of the lateral ventricles and abnormal increased signal in the deep periventricular white matter (arrows) bilaterally (a). Axial T1 FLAIR sequence demonstrating signal reduction in the deep periventricular white matter (arrows) together with a generalised reduction in white matter volume. There is mild dilatation of the occipital horns of the lateral ventricles (b)

Views of the cerebellum of case III2 (family 1), demonstrating normal left and right cerebellar hemispheres at age 7 years and significant right and left cerebellar hemisphere atrophy by age 14 years
Family 2
Following a normal pregnancy and delivery, signs of developmental delay were suspected in III2 (Fig. 4) when he was first admitted to hospital with severe chest infection at age 10 months. Examined by a paediatric neurologist aged 2 years, the findings were recorded as global developmental delay with marked axial hypotonia. He sat independently at age 2 years and walked at age 5. Epilepsy commenced at age 7, initially in the form of drop attacks, but later evolving to a more generalised pattern. At age 9 years, a further severe pneumococcal chest infection required hospitalisation and he required ventilation for 2 weeks. He did not regain ambulation following this episode. Clinical features, which were thought especially noteworthy on paediatric neurological evaluation, were tremulousness with orofacial dyskinesia. He died in his sleep at age 9 years of a presumed epileptic-related event.

The pedigree of family 2
His brother, III3, currently aged 7 years walked at about 20 months, but his general development had been a source of concern to his parents and paediatricians from earlier. While his general understanding is better than that of his brother, he has no words. Epilepsy has not been a feature in this boy. Typically he has three severe chest infections per annum, occasionally requiring hospital admission.
Radiological findings
As in the cases described above, both brothers show a reduction in the volume of white matter and also increased T2 and decreased T1 signal in the deep white matter and mild dilatation of the lateral ventricles particularly at level of atria and occipital horns. Creatine level is normal on spectroscopy in III2. The cerebellum was normal in both brothers at age 6 years but in III2, follow-up studies at age 9 years showed inferior atrophy (Fig. 5).

Sagittal views of the cerebellar hemispheres in case III2 (family 2), demonstrating progressive cerebellar hemisphere atrophy between age 6 and 9 years
Family 3
III1 presented with delayed speech at age 3 years (Fig. 6). The pregnancy had been normal but feeding had always been slow. He had walked at 19 months of age. However, examination at 3 years showed clear evidence of developmental delay. Head circumference was normal and facial appearance was unremarkable. Re-examined at age 12 years, no recognisable dysmorphic features were evident. However, he had no speech and had been hospitalised on several occasions for pneumonia. He had no history of seizures. III3 presented with delayed walking, not achieving this milestone until age 3 years. He had seizures, of non-specific form, from age 6 years and been treated on multiple occasions for respiratory tract infections. Examined at age 12 years, there were no dysmorphic features noted, but there were clinical signs of hyper-tonicity in the lower limbs. He had no speech. His younger brother III4 had walked at 2 years but examined at age 7, had no speech and had been hospitalised twice in the preceding 6 months with severe respiratory infections. There was no history of epilepsy.

The pedigree of family 3
Radiological findings
Enlargement of the fourth ventricle and destruction of the inferior aspects of the cerebellar hemispheres, more marked on the right (Fig. 7), were seen in an MRI scan taken at age 12 years in III1. MRI at age 12 in patient III3 showed a symmetrical increase in the T2 signal in the deep periventricular white matter posteriorly (Fig. 8) and no evidence of cerebellar destruction. MRI scan at age 7 years in III4 established similar findings to his older brother, with bilateral, symmetrical increased T2 signal of the deep periventricular white matter posteriorly (Fig. 9). No spectroscopic abnormality was identified in any patient.

Sagittal T1 FLAIR MR showing some destruction right inferior cerebellum (arrow; a) case III1 age 12 years. Axial T2 FLAIR brain in the same patient showing very small area of increased signal in the right occipital lobe (b)

Axial T2 FLAIR showing symmetrical increased T2 signal in the deep periventricular white matter posteriorly in case III3 at age 12 years

Patient III4 at age 7 years. Axial T2 FLAIR showing bilateral symmetrical increased T2 signal in the deep periventricular white matter posteriorly. There are small discrete areas of reduced signal in these areas in keeping with prominent Virchow–Robin spaces
Materials and methods
MLPA
The multiple ligation-dependent probe amplification (MLPA) P106 MRX kit, MLPA P015C kit and MLPA P049 SLC6A8 kit are developed and manufactured by MRC-Holland, Amsterdam, The Netherlands. Details on probe sequences can be found at the company’s website (http://www.mrc-holland.com). MLPA was performed according to the manufacturer’s protocol using 50 ng of genomic DNA per reaction. In all runs, DNA from at least two unrelated unaffected control persons was included. Reaction products were analysed on an ABI model 3100 capillary sequencer (Applied Biosystems, Denmark) using Genescan-ROX 500 standards and GeneMarker software to size the polymerase chain reaction (PCR) products and to obtain peak areas. Alterations were suspected if a relative peak area of a probe target sequence deviated more than 30% from the controls. The controls were matched with the sex of the patients.
X-inactivation analysis
For studying, the X-inactivation in the polymorphic androgen receptor locus 1 μg of genomic DNA was digested to completion with the methylation-sensitive enzyme HpaII and in parallel 1 μg of genomic DNA was incubated in the same buffer without enzyme. A fraction of the DNA (200 ng) was used for a PCR reacting using the upper-primer 5′-gcctgttgaactcttctgagc-3′ and the lower primer 5′-gctgtgaaggttgctgttcctc-3′. In order to be able to detect the PCR fragment, the upper primer was FAM-labelled. Reaction products were analysed on an ABI model 3100 capillary sequencer (Applied Biosystems, Denmark) using Genescan-ROX 500 standards and GeneMapper 3.0 software to size the PCR products and to obtain peak areas.
Genetic and biochemical investigations family 1
Karyotyping of both affected patients was undertaken several times and was normal. Maternal X-inactivation studies in the clinically and intellectually normal mothers of the probands II1 and II4 showed 100% unilateral X-inactivation. The duplicated region was clearly established in both mothers, in the surviving affected index case (III2) and in his more mildly affected sister III4 and extended at least from 152.6 to 153.3 Mb and includes the PNCK, SLC6A8, IDH3G, L1CAM, IRAK, MECP2 and GDI1 genes, according to the gene-specific probes present in the MLPA kits. From this result, we deduce that also the internally located genes ABCD1, FLNA and EMD are duplicated. A normal signal was obtained for the probe against the IDS gene (located at 148.4 Mb) and the probe against F8 (located at 153.8 Mb) flanking the duplication (Fig. 10). X-inactivation studies in III4 demonstrated skewed X-inactivation in the ratio of 70:30 and were presumed to account for the mild female phenotype. Biochemical investigation for creatine transport abnormalities was normal.

A fraction of the genes located at the X chromosome bands is shown. The location is obtained from Ensembl Homo sapiens version 53.36o (NCBI36). The full lines illustrate the genes known to be duplicated according to the MLPA probes. The hatch lines illustrate regions which might or might not be duplicated. The white box in the middle of the chromosome band illustrates a gap of several megabytes. In families 1 and 2, at least the region from 152.60 (SLC6A8) to 152.61 (PNCK) is duplicated. The flanking genes IDS (148.36) and F8 (153.71) are present in one copy at each X chromosome. In family 3, at least the region from 152.92 (IRAK1) to 152.94 (MECP2) is duplicated. The flanking genes LICAM (152.78) and GDI1 (153.31) are present in one copy at each X chromosome
Genetic and biochemical investigations family 2
Normal karyotyping was followed by normal sub-telomere studies, and it was a specific request for MECP/MRX2 studies which demonstrated the duplication of this locus within the localised region of Xq28 duplication. The duplication was confirmed in both affected brothers, with the same MLPA results as obtained for family1. Also in this family, the duplication extended at least from 152.6 to 153.3 Mb, including the PNCK, SLC6A8, ABCD1, IDH3G, L1CAM, IRAK, MECP2, FLNA, EMD and GDI1 genes with a normal signal for the probe against the IDS gene (located at 148.4 Mb) and the probe against F8 (located at 153.8 Mb) flanking the duplication (Fig. 10). Biochemical investigation for creatine transport abnormalities was normal.
Genetic investigations family 3
The duplication in this family is substantially smaller than the duplication in families 1 and 2 and includes IRAK (152.9), MECP2 and FLNA (153.2). Normal signal was obtained for the flanking genes L1CAM (152.8) and GDI1 (153.3; Fig. 10).
Discussion
Lubs et al. [6] were the first to draw attention to a condition of X-linked mental retardation associated with a propensity to multiple severe respiratory infections. These authors reported a family of five males, noting that three of the affected individuals had succumbed at ages 9, 8 and 5 years, respectively. In addition, they drew attention to the swallowing dysfunction, gastro-oesophageal reflux and hypotonia which they felt characterised the disorder and proceeded to map the condition to Xq28.
Mutations in the MECP2 gene were identified as causing Rett syndrome in 1999 [1]. Although variant clinical forms of the condition exist and can be associated with gross rearrangements of the gene [2, 5], most cases of Rett syndrome are female and pursue a classical clinical course with mental retardation and regression, stereotypic hand movements and regression of speech. However, missense mutations in the MECP2 gene have also been shown to be responsible for rare examples of males who die of severe neonatal encephalopathy in families with recurrent Rett syndrome [10].
Meins et al. [8] reported the first case of Xq28 duplication in a male patient with severe mental retardation and some features reminiscent of Rett syndrome. The subject of that report showed severe developmental delay, absence of speech and onset of epileptiform activity from age 6 years. This finding demonstrated, for the first time, that not only impaired or abolished gene function but also duplication and overexpression of MECP2 can cause mental retardation with features of Rett syndrome.
Further evidence of an emerging syndrome of Xq28 duplication involving the MECP2 locus came from Van Esch et al. [11]. Epilepsy, absence of speech, exaggerated deep tendon reflexes and a propensity to severe respiratory infections characterised the patients reported by these authors, family history sometimes revealing early neonatal deaths, among males from severe respiratory infections.
More recently, Frietz et al. [4] have recorded six pedigrees with multiple affected males showing severe mental retardation, hypotonia, recurrent respiratory infection and drawn attention to the value of the history of disproportionate propensity to recurrent chest infections, often associated with death, as an indicator of this underlying syndrome. They point out that many of the 18 cases described prior to their report had one or more episodes of mechanical ventilation, while tracheostomy was common among survivors. Further cases have been recorded by Echenne et al. [3], emphasising the progression from initial hypotonia to spasticity, the absence of language and the frequent association with myoclonic–astatic type seizures.
Lugtenberg et al. [7] present clinical and molecular details from six further families. Such neuroimaging details as were presented are inconsistent, findings ranging from large Virchow–Robin spaces, moderate cerebral atrophy, hyper-intensity of the signal from the posterior part of the cerebral white matter but with normal cerebellum and generalised ventricular dilatation. The authors acknowledge that it was not possible to determine a more specific cerebral phenotype but attest to the desirability of such a correlation in a new study.
We report here three new families with an Xq28 duplication. We describe seven boys with the symptoms frequently associated with Xq28 duplication: recurrent infections, hypotonia and mental retardation. Seizures are also common in the disorder, seen in 51% in the series of Lugtenberg et al. [7]. More novel, however, is our consistent finding, among all seven boys in our cohort, of a reduction in the volume of white matter, increased T2 and decreased T1 signal in the deep white matter and mild dilatation of the lateral ventricles. This represents a significant milestone in further delineating this syndrome. Moreover, the identification of progressive cerebellar changes among three older patients in our families, whose cerebellar imaging was initially normal but had later radiological evidence of atrophy, suggests that this is another important neuroradiological feature of this condition (Figs. 3, 5 and 7). We acknowledge that seizure-related activity can result in cerebral damage. The findings of cerebellar changes in case III1 of family 3 are especially noteworthy, as he did not have seizures and his cerebellar degenerative changes, also seen in patients III2 (family 1) and III2 (family 2) both of whom do have long-standing seizure disorders, are likely to indicate that this neuroradiological phenomenon is directly related to the duplication rather than to a secondary seizure-related effect.
Abnormal neuroradiological findings did not form part of the previously observed phenotypes, and published studies have been inconsistent in the presentation of such data—an understandable consequence of data unavailability. In addition to the observations of Lugtenberg et al. [7] in respect of the findings in their patients, outlined previously, Lubs et al. [6] provided neuroradiological data on some of their patients, indicating that one affected individual had mild cerebral atrophy on CT scan at age 8 years and recording mild atrophy in another brother at age 4 years. More significantly however, they reported post-mortem neuropathological findings in one individual in the family who died at age 5, specifically commenting that while the cerebral cortex was normal, ‘there was extensive Purkinje cell loss in the cerebellum.’ These authors also observed severe seizures and rapid central nervous system deterioration in some of the deceased individuals. Among Xq28 duplication cases reported by Echenne et al. [3], Frietz et al. [4], Meins et al. [8] and Van Esch et al. [11], some brain scan data have been reported, including non-specific white matter changes, delayed myelination, mild cortical atrophy and mild enlargement of the ventricles. White matter and ventricular dilatation such as we observe are recorded in some affected individuals, but there are no reports of follow-up studies in affected patients into the second decade of life. The cerebellar degenerative changes we observe in three of four patients having examination after the age of 9 years might not have been apparent in previously reported cases of this condition for this reason.
There are several genes known to be involved in mental retardation in the duplicated region in the reported literature. The identified duplications, ranging from 100 to 900 kb in size, are variable but always include the MECP2 and IRAK1 genes. The duplication we observe in families 1 and 2, who are not known to be related, is among the larger instances reported, although family K8300 of Frietz et al. [4] also had a duplication of comparable extent, as was also true of family F in the Lugtenberg et al. [7] report. Family 3 has a smaller duplication (Fig. 10). In common with other authors, we do not observe a discernible difference in phenotype between the different duplication sizes. Others have tried to delineate the possible contributions to phenotype which might be attributed to the different loci contained within the reported duplications but with little evidence of a linear relationship [7], and we do not attempt to add to these speculative discussions.
Lugtenberg et al. [7] report that the female relatives of the affected male cases in their series were all healthy, a finding which parallels previous reports of this condition. Our observations of the first manifesting carrier to date show that X-inactivation cannot be exclusively relied upon to spare the females who carry the duplication from the clinical ill effects. The previously reported cases demonstrated asymptomatic carrier females with extreme (>80%) or complete (100%) skewing of one of there X chromosomes. The present study indicates that a 70% skewing or less will lead to manifestation of the disease in females.
The overall prevalence of this condition is estimated to be 1% of unexplained X-linked mental retardation and maybe 2% of males with severe encephalopathy, but further large scale, studies in cohorts of severely mentally handicapped males are required for such estimates to be consolidated. Patients with mental retardation, recurrent infections, hypotonia, seizures and hyper-reflexia should be offered testing for Xq28 duplication. We suggest that recognition of the neuroradiological phenotype we describe will facilitate the identification of further cases
Abbreviations
- MLPA:
-
Multiple ligation-dependent probe amplification
- Array CGH:
-
Array comparative genome hybridisation
- Kb:
-
Kilobases
References
Amir RE, Van den Veyer IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188
Archer HL, Whatley SD, Evans JC, Ravine D, Huppke P, Kerr A (2006) Gross rearrangements of the MECP2 gene are found in both classical and atypical Rett syndrome patients. J Med Genet 43:451–456
Echenne B, Roubertie A, Lugtenberg D, Kleefstra T, Hamel BC, Van Bokhoven H, Lacombe D, Phillippe C, Jonveaux P, dr Brouwer AP (2009) Neurologic aspects of MECP2 gene duplication in male patients. Pediatr Neurol 41(3):187–191
Frietz MJ, Jones JR, Clarkson K, Lubs H, Abuelo D, Blaymore Bier J-A, Shashidhar P, Simensen R, Williams C, Giampietro PF, Schwartz CE, Stevenson R (2008) Recurrent infections, hypotonia and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics 118:1687–1695
Laccone F, Junemann I, Whatley S, Morgan R, Butler R, Huppke P, Ravine D (2004) Large deletions of the MECP2 gene detected by gene dosage analysis in patients with Rett syndrome. Hum Mutation 23:234–244
Lubs H, Abidi F, Blaymore Bier J-A, Abuelo D, Ouzts L, Voeller K, Fennell E, Stevenson RE, Schwartz CE, Arena F (1999) XLMR syndrome characterized by multiple respiratory infections, hypertelorism, severe CNS deterioration and early death localises to Xq28. Am J Med Genet 85:243–248
Lugtenberg D, Kleefstra T, Oudakker AR, Nillesen WM, Yntema HG, Tzschach A, Raynaud M, Rating D, Journel H, Chelly J, Goizet C, Lacombe D, Pedespan JM, Echenne B, Tariverdian G, O’Rourke D, King MD, van Kogelenberg M, Van Esch H, Gecz J, Hamel BC, van Bokhoven H, de Brouwer AP (2009) Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. Eur J Hum Genet 17:444–453
Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, Epplen JT (2005) Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet 42:e12
Raymond FL (2006) X linked mental retardation: a clinical guide. J Med Genet 43:193–2000
Schanen NC, Kurczynski TW, Brunelle D, Wodcock MM, Dure LS IV, Percy AK (1998) Neonatal encephalopathy in two boys in families with recurrent Rett syndrome. J Child Neurol 13:229–231
Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, Moraine C, Marynen P, Fryns JP, Froyen G (2005) Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 77:442–453
Conflicts of interest
All authors declare no conflict of interest regarding this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reardon, W., Donoghue, V., Murphy, AM. et al. Progressive cerebellar degenerative changes in the severe mental retardation syndrome caused by duplication of MECP2 and adjacent loci on Xq28. Eur J Pediatr 169, 941–949 (2010). https://doi.org/10.1007/s00431-010-1144-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-010-1144-4