
Abstract
Of the congenital disorder of glycosylation (CDG) syndromes, type 1a is the most common. CDG 1a is a multisystem disorder with a wide clinical spectrum. We report on a term newborn with a severe and fatal clinical course of CDG 1a syndrome. Skin fibroblasts showed a reduced activity of phosphomannomutase 2 (PMM2) and mutation analysis revealed a compound heterozygous PMM2gene mutation (F119L/F157S). Presenting features at birth were hypertrophic non-obstructive cardiomyopathy, “orange-peel” skin, inverted nipples and a hydrops-like aspect due to marked peripheral oedema. Suspected hydrops fetalis was not confirmed due to lack of ascites and pleural effusions. Striking clinical problems were therapy-resistant arterial hypertension, recurrent pericardial and pleural effusions and feeding difficulties with failure to thrive. Persistent congenital thrombocytopenia and hyperferritinaemia in the absence of infection were noted. Bone marrow cytology revealed a macrophage activation of unknown aetiology. Conclusion:Congenital thrombocytopenia, unspecific macrophage activation and a hydrops-like aspect without a real hydrops fetalis broaden the already wide phenotypic spectrum of congenital disorder of glycosylation syndrome type 1a.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Congenital Disorder of Glycosylation (CDG) syndromes are heterogeneous metabolic disorders of autosomal recessive inheritance, caused by defects in the glycoconjugation of proteins. As a result, they usually affect multiple organ systems. So far, 14 different CDG types have been characterised, each with a different enzymatic defect, accounting for approximately 80% of all CDG patients [8].
CDG-1a (McKusick 212065) is the most frequent and best characterised type with more than 500 reported patients. The underlying cause is a deficiency of phosphomannomutase 2 (PMM2), a cytosolic enzyme that converts mannose 6-phosphate into mannose 1-phosphate first characterised in 1995 [12]. Many mutations have been found in the corresponding PMM2gene [2,10]. Clinical features in the neonatal period include abnormal subcutaneous adipose tissue (fat pads, “orange peel” skin), inverted nipples, psychomotor retardation, cardiomyopathy and feeding problems with failure to thrive. In the first years of life, mortality is approximately 20% [6]. Here, we report a severe case of CDG-1a syndrome presenting typical features and some new clinical findings.
Case report
The patient, a girl, was the second child of healthy unrelated parents. Pregnancy was uneventful except for gestational diabetes treated by diet. The mother noticed normal limb movements during pregnancy and the girl was born at 40 weeks of gestation by caesarean section due to cessation of labour. At birth, weight was 4100 g (94th percentile), length was 51 cm (40th percentile) and head circumference was 35.5 cm (70th percentile). APGAR scores and pH of umbilical venous blood were normal.
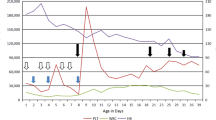
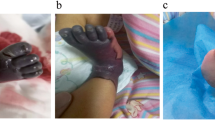
We saw a tachypnoeic newborn with a hydrops-like aspect due to marked peripheral oedema, in particular labial oedema (Fig. 1). Further features were an “orange peel” skin, inverted nipples, multiple haematomas and petechiae. Neurological examination revealed a weak sucking reflex, absence of Moro reflex, muscular hypotonia and hyperexcitability. Apart from the skin appearance, we could not confirm the suspected diagnosis of “hydrops fetalis” as ultrasound did not reveal ascites or pleural effusions. Echocardiography demonstrated non obstructive hypertrophic cardiomyopathy (right and left ventricle) with a pericardial effusion of 4–6 mm, dilatation of the right ventricle and myocardial dysfunction. A chest radiograph showed mild respiratory distress syndrome. Laboratory data on admission are given in Table 1. Severe thrombocytopenia persisted throughout the clinical course with minimal platelet counts of 9×109/l and a maximum of 66×109/l. There was no evidence of bacterial or viral infection. Several blood cultures were negative; serum markers for inflammation were not elevated. Specific antibody titres for CMV, EBV, herpes simplex, parvo B 19, picorna-virus and toxoplasmosis were negative. On the 15th day of life, initial thrombocytopenia was followed by total bone marrow suppression: haematocrit 21%, leukocytes 3.3×109/l, platelets 9×109/l. Leukocyte count normalised, but anaemia and thrombocytopenia persisted. Extremely high ferritin levels were measured from the 5th to the 8th week of life with a maximum of 8819 ng/ml (reference range 15–120 ng/ml).

The patient on the 5th day of life still requiring continuous positive airway pressure ventilation. Prominent are marked peripheral oedema, especially labial oedema, and an “orange peel” skin
From the 1st day of life, arterial hypertension was present with systolic blood pressures above 100 mmHg, persisting despite antihypertensive treatment with beta-blockers, diuretics and ACE-inhibitors. Myocardial function could not be improved and pericardial effusion increased. Finally, drainage of pericardial effusions and pleural effusions was required. Moreover, severe clinical problems were feeding difficulties with failure to thrive. The patient tolerated only small feeding volumes and vomited frequently and finally required gastroduodenal tube feeding. Stools were loose and watery. During the course of the disease, respiratory and myocardial function deteriorated and the girl died after 8 weeks of life.
Post-mortem findings showed hypertrophic non-obstructive cardiomyopathy, enlarged polycystic kidneys, incomplete liver cirrhosis, pancreas fibrosis, ascites, and pericardial and pleural effusions. Neuropathology revealed partial cerebellar atrophy, especially of the vermis, and generalised white matter gliosis. Bone marrow cytology showed macrophage activation with phagocytosis of erythropoetic elements. There were no histopathological signs of viral or mycobacterial infection in any organ.
Finally, the clinical aspect with “orange peel” skin, inverted nipples and cardiomyopathy led to CDG syndrome as the potential underlying disease. Isoelectric focussing of serum transferrin performed on the 50th day demonstrated a characteristic pattern of CDG syndrome. The specific enzyme activity of PMM2 in skin fibroblasts was reduced to 0.13 mU/mg protein (control: 2.9 mU/mg protein), confirming the diagnosis of CDG-1a. The activity of phosphomannose isomerase was normal (10.5 mU/mg protein, control: 10.2 mU/mg protein). Genomic analysis revealed a compound heterozygous F119L/F157S mutation in the PMM2gene. The parents refused genotyping.
Discussion
Our patient presented the typical clinical features of CDG-1a syndrome such as hypertrophic cardiomyopathy, inverted nipples and “orange peel” skin. However, we found several clinical and laboratory findings that have not been reported so far in patients with CDG-1a syndrome. In detail, we found a hydrops-like skin appearance without ascites or pleural effusions, a congenital persistent thrombocytopenia and finally extremely elevated ferritin levels due to an unspecific macrophage activation.
Until now, a non-immune hydrops fetalis has been reported to be associated with CDG-1 syndrome. Originally, this was based on a single case report of two siblings and the hydrops was described clinically without major criteria such as ascites, pleural or pericardial effusions being mentioned [4]. In our case, we noticed marked peripheral oedema as in true hydrops fetalis; however, major criteria such as ascites or pleural effusions were absent. Therefore, we question if there is an association between CDG-1 syndrome and hydrops fetalis and the clinical term “hydrops” should be used with caution. In our case, cardiomyopathy could explain the pericardial effusion but not skin oedema without ascites or pleural effusions. The skin oedema improved over time but the “orange peel” skin still remained (Fig. 2).

The patient at 4 weeks of age with improved peripheral oedema but still retaining an “orange peel” skin
The aetiology of the persistent and severe thrombocytopenia and bone marrow suppression on day 15 remains unexplained. A viral infection could be the underlying course, but we excluded the most likely ones. Also a neonatal allo-immune thrombocytopenia was ruled out. A disseminated intravascular coagulopathy was not likely, both clinically and according to laboratory data (Table 1). Elevated d-dimers are a known coagulation abnormality found in CDG-1 syndrome [5]. Until now, two siblings with CDG-1 and severe thrombocytopenia have been reported, but in vitro testing of skin fibroblasts showed normal activity of both PPM2 and phosphomannose isomerase [1].
Extremely elevated ferritin levels as in our patient have not been reported in CDG patients so far. The post-mortem bone marrow cytology showed macrophage activation, but did not fulfill all criteria for a haemophagocytic syndrome. Typical features like splenomegaly, lymphadenopathy, elevated triglycerides, low fibrinogen and elevated liver enzymes were absent [11]. Finally, this unspecific macrophage activation could explain the hyperferritinaemia. A primary association with CDG-1a syndrome is possible, but also viral infection (we could not specify) or multiple organ failure could account for the unspecific macrophage activation.
The mutation analysis revealed a compound heterozygous state of two known mutations F119L/F157S on both PMM2alleles. The F119L mutation accounts for approximately 16% of all mutant alleles and is common [10]. The F157S mutation is rare and has been reported in a Spanish child with a compound F157S/D65Y mutation, severe neonatal presentation, and early death [3]. Our child with the genotype F119L/F157S was severely affected and died after 8 weeks of life. Therefore, a correlation between the F157S mutation and a severe neonatal phenotype might be possible. This hypothesis is further supported by homozygous cases of either D65Y or F119L presenting a less severe phenotype [7,9]. Finally, for a more detailed genotype phenotype correlation, more cases need to be collected and evaluated in a systematic manner. Furthermore, to support genotyping, detailed assessment and description of clinical abnormalities are needed.
Abbreviations
- CDG :
-
congenital disorder of glycosylation
- PMM2 :
-
phosphomannomutase 2
References
Acarregui MJ, George TN, Rhead WJ (1998) Carbohydrate-deficient glycoprotein syndrome type 1 with profound thrombocytopenia and normal phosphomannomutase and phosphomannose isomerase activities. J Pediatr 133: 697–700
Bjursell C, Erlandson A, Nordling M, Nilsson S, Wahlström J, Stibler H, Kristiansson B, Martinsson T (2000) PMM2 mutation spectrum, including 10 novel mutations, in a large CDG type Ia family material with focus on Scandinavian families. Hum Mutat 16: 395–400
Briones P, Vilaseca MA, Schollen E, Ferrer I, Maties M, Busquets C, Artuch R, Gort L, Marco M, van Schaftingen E, Matthijs G, Jaeken J, Chabas A (2002) Biochemical and molecular studies in 26 Spanish patients with congenital disorder of glycosylation type Ia. J Inherit Metab Dis 25: 635–646
De Koning TJ, Toet M, Dorland L, de Vries LS, van den Berg IE, Duran M, Poll-The BT (1998) Recurrent nonimmune hydrops fetalis associated with carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 21: 681–682
Fiumara A, Barone R, Buttitta P, Musso R, Pavone L, Nigro F, Jaeken J (1996) Haemostatic studies in carbohydrate-deficient glycoprotein syndrome type I. Thromb Haemost 76: 502–504
Jaeken J, Carchon H (1993) The carbohydrate-deficient glycoprotein syndromes: an overview. J Inherit Metab Dis 16: 813–820
Kjaergaard S, Schwartz M, Skovby F (2001) Congenital disorder of glycosylation type Ia (CDG-Ia): phenotypic spectrum of the R141H/F119L genotype. Arch Dis Child 85: 236–239
Marquardt T, Denecke J (2003) Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr 162: 359–379
Matthijs G, Schollen E, Van Schaftingen E, Cassiman JJ, Jaeken J (1998) Lack of homozygotes for the most frequent disease allele in carbohydrate-deficient glycoprotein syndrome type Ia. Am J Hum Genet 62: 542–550
Matthijs G, Schollen E, Bjursell C, Erlandson A, Freeze H, Imtiaz F, Kjaergaard S, Martinsson T, Schwartz M, Seta N, Vuillaumier-Barrot S, Westphal V, Winchester B (2000) Mutations in PMM2 that cause congenital disorders of glycosylation type Ia (CDG Ia). Hum Mutat 16: 386–394
Sawhney S, Woo P, Murray KJ (2001) Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child 85: 421–426
Van Schaftingen E, Jaeken J (1995) Phosphomannomutase deficiency is a cause of carbohydrate-deficient glycoprotein syndrome type I. FEBS Lett 377: 318–320
Acknowledgements
We thank Prof. Christian Körner, University of Heidelberg, Germany, for kindly performing the enzymatic phosphomannomutase test and Drs. D. Haas, V. Peters and A. Schulze, University of Heidelberg, Germany, for performing the isoelectric focusing of serum transferrin. We kindly acknowledge Dr. Poske, PD Dr. Horn, Prof. Schober and Prof. Wittekind, Dept. of Pathology, University of Leipzig, for performing the autopsy and bone marrow cytology. Finally we thank Dr. Klemens Raile, University Children’s Hospital Leipzig, for detailed work on the manuscript and critical discussion of this case.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Noelle, V., Knuepfer, M., Pulzer, F. et al. Unusual presentation of congenital disorder of glycosylation type 1a: congenital persistent thrombocytopenia, hypertrophic cardiomyopathy and hydrops-like aspect due to marked peripheral oedema. Eur J Pediatr 164, 223–226 (2005). https://doi.org/10.1007/s00431-004-1611-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-004-1611-x