
Abstract
Local innate immunity plays a key role in initiating and coordinating homeostatic and defense responses in the heart. We have previously reported that the cardiotropic parasite Trypanosoma cruzi, the etiological agent of Chagas disease, protects cardiomyocytes against growth factor deprivation-induced apoptosis. In this study, we investigated cardiomyocyte innate immune response to T. cruzi infection and its role in cellular protection from apoptosis. We found that Toll-like receptor (TLR) 2-expressing cells were strongly increased by the parasite in BALB/c neonatal mouse cardiomyocyte cultures. Using a dominant-negative system, we showed that TLR2 mediated cardiomyocyte survival and the secretion of interleukin (IL) 6, which acted as an essential anti-apoptotic factor. Moreover, IL6 released by infected cells, as well as the recombinant bioactive cytokine, induced the phosphorylation of the signal transducers and activators of transcription-3 (STAT3) in cultured cardiomyocytes. In accord with the in vitro results, during the acute phase of the infection, TLR2 expression increased 2.9-fold and the anti-apoptotic factor Bcl-2 increased 4.5-fold in the cardiac tissue. We have clearly shown a cross-talk between the intrinsic innate response of cardiomyocytes and the pro-survival effect evoked by the parasite.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the mechanisms by which the innate immune system senses the presence of foreign antigens is through TLRs, which recognize the pathogen-associated molecular patterns that are shared by different pathogen groups [1]. The stimulation of these receptors leads to transcriptional regulator nuclear factor kappa B (NF-κB) activation and cytokine production. Pro-inflammatory cytokines, in turn, appear to play a central role in the orchestration and timing of the intrinsic cardiac stress response, by providing instantaneous anti-apoptotic cytoprotective signals that allow tissue repair and/or remodeling. Cytokines of the IL6 family, expressed by all myocardial cell types, are involved in the inhibition of cardiomyocyte apoptosis and hypertrophy [2, 3]. Although high levels of IL6 have been detected following infection with the cardiotropic parasite Trypanosoma cruzi [4, 5], little is known about the consequences of IL6 production on cardiac cells.
Trypanosoma cruzi is an intracellular protozoan which causes Chagas disease. Endemic to several regions in Latin America, this disease persists as the major infectious heart disease worldwide [6], with a global incidence of 300,000 new cases per year. Chagas disease can be characterized by two distinct periods [7], an acute phase and a chronic one. During the acute phase, which lasts 2–4 months, the parasite can be detected in the blood stream as well as in the host tissues. This stage is followed by a life-long chronic phase, in which parasites are effectively cleared from most tissues but persist for years in cardiac cells. Nowadays, it is widely accepted that the persistence of parasites in the myocardium is an important factor in the development of Chagas disease. Thus, the identification of the mechanisms that enable the parasite to evade the host immune response and to survive within the target cells is crucial in order to understand the pathogenesis of the disease.
Similar to other intracellular parasites, T. cruzi invades and resides inside host cells, thereby avoiding direct destruction by the immune system. The infected cell, however, has the capacity to initiate its own death by apoptosis. This potent defense mechanism exerts strong selective pressure on T. cruzi, which has evolved strategies to modulate the target cell apoptotic program in its favor [8–11]. Related to this, the first study showing that T. cruzi infection promotes the prolonged survival of cardiomyocytes was reported by our group [12]. We found that T. cruzi infection protects isolated cardiac cells from apoptosis induced by growth factor deprivation by increasing the expression of the anti-apoptotic factor Bcl-2. The protection was improved by pretreatment of the target cells with cruzipain (the major cysteine protease of T. cruzi) devoid of enzymatic activity [12]. Furthermore, we reported that at least two signal transduction pathways are activated in cardiomyocytes exposed to T. cruzi tripomastigotes, the PI3K/Akt and MEK1/ERK, with both pathways leading to a decrease in activated caspase-3 [13].
In this study, we investigated the functional relation between innate recognition of T. cruzi by cardiac cells and its possible role in target cell protection against apoptosis. The implications of the findings for the pathogenesis of the disease are discussed.
Materials and methods
Animals and infection
BALB/c and C57BL/6 mice were purchased from the Facultad de Ciencias Veterinarias, Universidad Nacional de La Plata, Argentina. Animals were maintained at the Animal Resource Facility of the CIBICI-CONICET (National Institutes of Health-USA assurance number A5802-01) in agreement with the Guide to the Care and Use of Experimental Animals published by the Canadian Council on Animal Care and approved by the CIBICI-CONICET committee. C57BL/6 TLR2-knockout mice (from The Jackson Laboratory, Bar Harbor, ME, USA) were maintained at the Animal Resource Facility of the Medical School of Ribeirão Preto (Universidade de Sao Paulo, Brasil). Six-week-old BALB/c females were infected i.p. with 1 × 103 T. cruzi blood-derived tripomastigotes of Tulahuen strain. Bloodstream tripomastigotes forms were harvested by heart puncture from T. cruzi-infected BALB/c mice at the peak of parasitemia. Noninfected mice were used as control. Parasites were maintained by serial passages from mouse to mouse.
Neonatal mouse primary cardiomyocyte culture
Primary cultures of cardiomyocytes from neonatal BALB/c, C57BL/6 and C57BL/6 TLR2-knockout mice were performed as described previously [12]. For selective enrichment of cardiac myocytes, cell suspensions were preplated for 2 h, during which time the nonmyocytes readily attached to the bottom of the flask. The cells remaining in the supernatant were then collected and plated at a density of 7 × 104 cells/cm2 on gelatin/fibronectin-coated 24-well plates in 10% FBS-DMEM. The cultures were kept in a 5% CO2 incubator at 37°C to allow the cells to adhere firmly to the multiwell and start beating. More than 85% of cells were cardiomyocytes as detected by immunostaining with antibody to mAchR M2. After 24 h, the cells were washed and infected with different doses of Tulahuen T. cruzi tripomastigotes, obtained from the supernatant of infected LLC-MK2 cells (a cell line from kidney adult Rhesus monkey). After 3 h, the monolayers were washed to remove the excess of parasites. Nevertheless, several tripomastigotes remained attached to the monolayers. The cultures were maintained in complete culture medium or in 0.1% FBS-DMEM. When indicated in the legend of the figure, infected monolayers were treated with 25 μM N-α-tosyl-l-phenylalanine chloromethyl ketone (TPCK) (Sigma-Aldrich), purified rat anti-mouse IL6 (5 μg/ml) or IgG1 isotype control (5 μg/ml) (both from BD Pharmingen). Other cultures were stimulated with or without biologically active recombinant mouse IL6 (5 ng/ml) (eBioscience), or the TLR2 ligand PAM3CSK4 (0.5 μg/ml) (Invivogen).
Cardiomyocyte transfection
Neonatal mouse cardiac cells were plated in complete media at a density of 6.5 × 104 cells/cm2 on gelatin/fibronectin-coated 24-well plates and cultured as described previously. After 2 days of culture, contracting cardiomyocytes were transfected with dominant negative forms of TLR2 (pZERO-mTLR2) (0.5 μg/well) or the empty control vector (pZERO-mcs) (0.5 μg/well) (both from Invivogen) using Lipofectamine Plus Reagent (Invitrogen), following the protocol of the manufacturer. Twenty-four hours after transfection, the cells were infected (cell/parasite ratio 1:2) and maintained in complete media for 3 h. Then, control, infected and PAM3CSK4 (0.5 μg/ml) (PAM3)-treated monolayers were switched to starved medium for 48 h. Apoptosis was measured by Annexin V-FITC labeling and flow cytometry analysis. The supernatants were collected for cytokine detection by ELISA, and the transfection efficiency was monitored by flow cytometry as illustrated in Fig. 2b.
Measurement of apoptotic cell death
Following T. cruzi infection or other treatments, the cardiomyocyte monolayers were washed once with cold PBS and incubated with the binding buffer containing 5 μl of FITC-Annexin V (BD Pharmingen) for 15 min on ice. Previous to acquisition, the cells were washed and stained with propidium iodide. A minimum of 30,000 events for each condition were analyzed by flow cytometry (FACSCanto II, Becton–Dickinson). Cellular debris, necrotic and late apoptotic cells were excluded from the analysis. Data were analyzed using FlowJo software 5.7.2 (Tree Star, Inc.).
Immunofluorescence
Heart specimens were fixed in 4% paraformaldehyde-PBS and embedded in paraffin. Deparaffinized and rehydrated sections were permeabilized and incubated with rabbit polyclonal anti-TLR2 (Santa Cruz Biotechnology) or rabbit polyclonal anti-Bcl-2 antibody (BD Pharmingen). After washing, these sections were incubated with Alexa Fluor 594 or with Alexa Fluor 488-conjugated anti-rabbit IgG, respectively (both from molecular probes), and nuclei were labeled using DNA-binding fluorochrome Hoechst or propidium iodide. The slides were observed and photographed using a FV300 Olympus confocal scanning microscope.
Flow cytometric analysis
For TLR2, TLR4 and mAchR detection, 0.2 × 106 cells were stained using a standard protocol with the following antibodies: goat polyclonal anti-mAchR antibody (Santa Cruz Biotechnology) followed by anti-goat-FITC IgG (eBioscience) together with PE-labeled monoclonal anti-mouse TLR2 (eBioscience) or with Alexa Fluor 488-labeled monoclonal anti-mouse-TLR4 antibody (eBioscience). Stained samples were acquired using the FACSCanto II cytometer (Becton–Dickinson), and data were analyzed using FlowJo software 5.7.2 (Tree Star, Inc.).
Cytokine assays
The ELISA was performed for the detection of IL1β, TNFα, IL12, IL6, IL17 and IL10 cytokines. Briefly, ELISA plates were coated with anti-cytokine antibodies (BD Pharmingen and e-Bioscience) overnight at 4°C. Then, these were washed and blocked with 10% FBS. Culture supernatants, obtained from cultures infected for 4, 8, 16 or 48 h, were incubated overnight at 4°C. Then, the plates were incubated with biotinylated anti-cytokine antibody (BD Pharmingen and e-Bioscience) for 1 h at room temperature. After washing, streptavidin-peroxidase (BD Pharmingen) was added and incubated for 30 min. The reaction was revealed using DakoCytomation TMB Substrate Chromogen (Dako), before being read at 490 nm in a Microplate reader (Bio-Rad). Standard curves were generated using recombinant cytokines (BD Pharmingen).
Western blot assays
Heart tissues were lysated for 30 min on ice. Isolated cultured myocytes were washed, scraped and mixed with loading buffer. Aliquots with equal amounts of protein were separated on SDS–PAGE and electrotransferred to nitrocellulose membranes (Bio-Rad Laboratories). After being blocked, the membranes were incubated with rabbit anti-TLR2, rabbit anti-Bcl-2 or with rabbit anti-Phospho-STAT3 antibody (Cell Signaling). After washing, the membranes were incubated with HRP-conjugated anti-rabbit IgG (Sigma) and assayed by the ECL chemiluminescent system (Amersham Pharmacia Biotech). The membranes for TLRs or STAT3 were stripped and incubated with anti-actin antibody or anti-STAT3 antibody (Cell Signaling), respectively, before being washed and revealed. The band intensity for TLR2, Bcl-2 or p-STAT3 was semi-quantified by densitometric scanning and normalized to that of actin or total STAT3, respectively, using ImageJ processing software (www.rsb.info.nih.gov/ij).
Statistical analysis
To compare different experimental conditions, an analysis of variance (two-way or one-way ANOVA) with the Bonferroni post hoc test was performed. A two-tailed Student′s t test was used for comparison between control and experimental samples. A P value <0.05 was considered significant (* P < 0.05; ** P < 0.005; *** P < 0.001, NS nonsignificant).
Results
Trypanosoma cruzi infection increases the percentage of TLR2-expressing cells in cultured cardiomyocytes
The heart expresses at least six TLRs; namely, TLR2, TLR3, TLR4, TLR5, TLR7 and TLR9, of which, the most studied are TLR2 and TLR4. Therefore, we first sought to examine the basal expression of TLR2 and TLR4 on isolated cardiomyocytes and their capacity to modulate the expression of these receptors in response to T. cruzi infection. We evaluated by flow cytometry the cell surface expression of TLR2 and TLR4 in cultured neonatal mouse cardiac myocytes at 0, 3, 6, 16 and 24 h post-infection and compared to uninfected controls. We found that cardiomyocytes expressed low constitutive levels of TLR2 and TLR4 (Fig. 1). Nevertheless, as early as 16 h, the infection significantly increased the percentage of TLR2-expressing cells, which were sustained for at least 24 h post-infection (Fig. 1a). Moreover, the increased expression was independent of the parasite dose (Fig. 1b). In contrast, the percentage of TLR4 positive cells was not significantly modified during the culture period, either in infected or uninfected cultures (Fig. 1a). Even though about 40% of cardiomyocytes expressed high levels of TLR2 24 h after infection, only 12 ± 5% of the cells were infected at this time point. This suggested that uninfected cells also displayed TLR2 expression. We observed that TLR2 expression was restricted to cardiomyocytes, since the great majority of TLR2-positive cells were also positive for M2 muscarinic receptor (mAchR), a marker of cardiomyocytes (Fig. 1c). These results rule out the possibility that other cell types present in cardiomyocyte primary cultures, such as fibroblasts, could be overexpressing TLR2.

Trypanosoma cruzi increases TLR2-expressing cells in cardiomyocyte cultures. Primary cardiomyocyte cultures were left untreated (Control) or infected with T. cruzi tripomastigotes at a cell/parasite ratio of 1:2 (T. cruzi). The gray line represents the isotype control. a Representative analysis of TLR2 and TLR4 expressing cells measured at 16 and 24 h post-infection. The bars represent mean ± SEM of three independent experiments. b The monolayers were infected with the indicated cell/parasite ratio, and TLR2 expression was measured 16 h later (black line) with control cultures being maintained in medium alone (gray line). c Infected cultures were stained for TLR2 and mAchR, a marker of cardiomyocytes. The contour plot shows that cells gated on the expression of TLR2 were also positive for mAchR. The histograms and contour plot are representatives of three separate experiments, performed in quadruplicate
TLR2-signaling is involved in the cardiomyocyte protection induced by Trypanosoma cruzi
TLR2 has been associated with a protective role in cardiomyocytes [14], with the activation of this receptor attenuating the apoptotic effects of ischemia/reperfusion in the myocardium [15]. It has also been reported that several surface glycoconjugates shed by infective T. cruzi tripomastigotes activate TLR2 in macrophages [16]. Thus, we set out to investigate the potential role of TLR2 in the parasite-induced cytoprotective effect.
In order to test the involvement of TLR2, primary cultures were transfected with a dominant-negative TLR2 (dnTLR2) plasmid or its empty control vector, followed by incubation with T. cruzi or the TLR2 agonist PAM3CSK4. We have previously shown that the death of cultured cardiomyocytes induced by serum-deprivation is related to apoptosis, as characterized by the appearance of apoptotic nuclei, positivity in TUNEL assays, hypodiploid nuclei stained with propidium iodide and Annexin V labeling [12, 13]. Therefore, we used Annexin V labeling here as a read out of apoptosis. As shown in Fig. 2a, the parasites and TLR2 ligand incubation decreased the apoptotic rate in cultures transfected with control vector. In contrast, overexpression of dnTLR2 protein blocked the cytoprotective effect induced by the parasite and by PAM3CSK4 (Fig. 2a). These results strongly suggest that the triggering of TLR2 is important for the induction of the cardiomyocyte survival observed in our model. Transfection efficiency was monitored by flow cytometric analysis of TLR2 expression. As shown in Fig. 2b, the expression levels were only up-regulated when the cells were transfected with dnTLR2 plasmid but not with the control vector plasmid, in comparison with nontransfected cells or mock-transfected controls.

Cardiomyocyte cytoprotection induced by T. cruzi involves signaling through TLR2. a Primary myocyte cultures were transiently transfected with dnTLR2 plasmid or the control vector. After 24 h, control, infected (cell/parasite ratio 1:2) and PAM3CSK4 (PAM3)-treated monolayers were switched to medium with 0.1% FBS for 48 h. Apoptosis was measured by flow cytometric analysis of Annexin V-stained cells. Results are expressed as means of quadruplicate wells ± SEM. Representative data of four independent experiments are shown. b Transfection efficiency was monitored by measuring TLR2 expression by flow cytometry 24 h after transfection with dnTLR2 plasmid (dnTLR2) or with the empty control vector (Vector). Other cultures were processed as mock-transfected controls (Mock) or maintained in medium alone (Control). The gray line represents the isotype control. Data are means of geometric means of fluorescence intensity (GMFI) ± SEM. Results are representative of two independent experiments performed in quadruplicate. c Cardiomyocyte cultures from TLR2-knockout neonatal mice were infected (T. cruzi) or maintained in medium (Control). After 48 h of serum starvation, apoptosis was measured by Annexin V staining. Data are representative of two independent experiments performed in quadruplicate
To further corroborate the involvement of TLR2 in the survival effect observed, we performed experiments with cardiac cells obtained from C57BL/6 TLR2-knockout mice. In these cultures, the anti-apoptotic effect of the infection was abolished (Fig. 2c). It is important to stress that T. cruzi also protected the cardiomyocytes obtained from wild-type C57BL/6 genetic background mice (8.9 ± 0.6% infected vs. 23.7 ± 2.6% medium, P < 0.005). Taken together, these results show that TLR2 signaling would be involved in the cytoprotective effect induced by T. cruzi, which is independent of the mouse genetic background tested.
Trypanosoma cruzi stimulates IL6 release by cardiomyocyte cultures
Since signaling through TLRs is primarily responsible for the production of pro-inflammatory cytokine by macrophages following exposure to T. cruzi, we analyzed the profile of cytokines produced early as a consequence of parasite interactions with cardiomyocytes, with 16 h-culture supernatants being screened for the presence of IL1β, TNFα, IL12, IL6, IL17 and IL10. Among the cytokines tested, we did not detect any significant production at earlier post-infection times (data not shown). We could only detect a sustained production of IL6 as from 16 h post-infection (Fig. 3). IL6 release was blocked by preincubation with TPCK, a compound shown to inhibit NF-κB transcriptional activity [17]. These findings illustrate that cardiomyocytes are able to efficiently respond to T. cruzi by producing IL6, in a manner dependent on NF-κB activation.

Trypanosoma cruzi stimulates IL6 release by cardiomyocytes. Primary cardiomyocyte cultures were left untreated or infected (cell/parasite ratio 1:2). Other infected monolayers were incubated with TPCK. The IL1β, TNFα, IL12, IL6, IL17 and IL10 cytokine production were quantified by ELISA in the 16 h supernatants. Data represent mean ± SEM of six independent experiments performed in triplicate
IL6 mediates the phosphorylation of STAT3 and the survival of cardiomyocytes
IL6 is a pleiotropic cytokine that modulates a variety of physiological events. Of relevance to our work, IL6 has been reported to exert a potent anti-apoptotic effect on cardiomyocytes [2, 18]. We, therefore, sought to test whether the IL6 secreted as result of exposure to T. cruzi played a role in the cytoprotection of cardiomyocytes. Depletion of IL6 by neutralizing antibodies in T. cruzi-infected cultures was found to eliminate the parasite-induced cytoprotection (Fig. 4a), whereas the incubation of infected monolayers with control IgG did not show any effect on the survival rate. The correlation between IL6 and cardiomyocyte survival was further corroborated by the incubation of serum-starved cell cultures with the bioactive recombinant cytokine (Fig. 4a). As control, Fig. 4b shows that IL6 was almost undetectable in the supernatant of cultures treated with the neutralizing antibodies. Then, we investigated whether T. cruzi-induced IL6 secretion occurred as a result of TLR2 activation by using cardiomyocytes transfected with dnTLR2. The parasite induced significant levels of IL6 in cells transfected with control vector, but these levels were lower in cells bearing the dnTLR2 construct (Fig. 4c). These results show a direct correlation between the activation of TLR2 and IL6 production induced during T. cruzi infection.

Trypanosoma cruzi-induced IL6 mediates the survival of cardiomyocytes. a The infected (cell/parasite ratio 1:2) monolayers were incubated with anti-mouse IL6 IgG (anti-IL6). Uninfected cultures were treated with recombinant IL6 (IL6) or maintained in medium alone (Control). All the cultures were shifted to serum starvation conditions for 48 h. b The supernatants were tested for IL6 production by ELISA. c Transiently transfected cardiac myocytes were incubated with medium alone or infected with tripomastigotes (cell/parasite ratio 1:2) in complete medium. Control or infected monolayers were switched to starved medium. The supernatants were collected after 48 h of culture for IL6 cytokine analysis. Results are expressed as means of quadruplicate wells ± SEM. Representative data of four independent experiments are shown. d Cardiomyocyte monolayers were incubated with the supernatants of infected cultures (T. cruzi), cultures infected and treated with blocking antibody (T. cruzi + anti-IL6), cultures infected and treated with an unrelated control IgG (T. cruzi + IgG1), or noninfected and incubated with bioactive recombinant IL6 cytokine (IL6) for 15 min. STAT3 phosphorylation was estimated by immunoblotting with anti-phospho-STAT3 (p-STAT3) or anti-STAT3 antibodies. The band intensity for p-STAT3 was normalized to that of total STAT3 (mean ± SEM)
In the heart, IL6 transduces its signals via the glycoprotein-130 receptor, predominantly to STAT3 [19]. Upon activation, STAT3 proteins become phosphorylated on a specific tyrosine residue (Tyr705) and undergo dimerization and translocation into the nucleus, where they regulate the expression of pro-survival genes [18]. Given the above results, we next investigated whether IL6 released in response to parasite incubation could activate the STAT3 pathway in primary cultures. While the 48 h supernatants from T. cruzi-infected monolayers dramatically increased the STAT3 phosphorylation 15 min after stimulation (Fig. 4d, T. cruzi), the supernatants of cultures treated with anti-IL6 blocking antibody (Fig. 4d, T. cruzi+anti-IL6), but not with the IgG control (Fig. 4d, T. cruzi+IgG1), failed to induce phospho-STAT3. As expected, the bioactive recombinant IL6, at the concentration produced by infected monolayers, also stimulated the phosphorylation of STAT3 (Fig. 4d, IL6). Collectively, these data strongly suggest that IL6 induced by the parasite and/or by parasite-derived component(s) activate the STAT3 signal transduction pathway in cardiomyocytes.
TLR2 and Bcl-2 expression are increased in cardiac tissues of mice infected with Trypanosoma cruzi
Finally, in order to know whether the infection was able to modulate the expression of TLR2 and the anti-apoptotic factor Bcl-2 in cardiac cells in vivo, we evaluated the cardiac expression of TLR2 and Bcl-2 in BALB/c mice during the course of the acute phase of the infection. In agreement with the in vitro findings, the infection led to an enhancement of the TLR2 and Bcl-2 expression. The immunoblotting of tissue lysates revealed with anti-TLR2 antibody showed that although this receptor was present in the heart tissue of control mice, its levels were significantly increased as early as 7 days post-infection (Fig. 5a). To determine the cellular source of TLR2 detected in the infected hearts, we performed immunofluorescence analysis. Confocal images in Fig. 5b show that infiltrating cells (Fig. 5b, middle) as well as cardiac fibers (Fig. 5b, right) stained positive for TLR2 in infected myocardium compared to the weak labeling observed in control samples (Fig. 5b, left). On the other hand, Bcl-2 expression was almost undetectable in normal tissue but was strongly increased after infection (Fig. 6). Confocal studies revealed that Bcl-2 immunoreactivity was predominantly localized in cardiomyocytes of the infected myocardium (Fig. 6b, right), but not in cardiac cells from normal mice (Fig. 6b, left). These results suggest that endogenous inhibitors of apoptotic cell death, such as Bcl-2, and the innate recognition receptors, are highly expressed in the infected myocardium, which may contribute to cell survival.

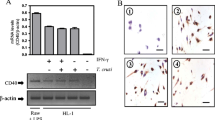
Increased expression of TLR2 in the heart of infected BALB/c mice. a Western blot analysis of TLR2 protein expression in heart tissue from mice injected with the vehicle alone (Control) or infected with 1 × 103 T. cruzi blood-derived tripomastigotes, at the indicated days post-infection (dpi). b Immunofluorescence staining for TLR2 of heart sections from control and infected mice. Arrows and arrowheads indicate infiltrating and myocyte cells, respectively. Slides were observed and photographed using a Zeiss LSM Pascal confocal scanning microscope. Six animals per group were analyzed at different dpi. The results are expressed as mean ± SEM of all samples analyzed, with each P value being determined versus Control. Scale bar = 20 μm

Increased heart expression of Bcl-2 during acute infection of BALB/c mice. a Western blot analysis of Bcl-2 at indicated dpi with samples from each time shown in duplicate (except for 24 dpi). b Immunofluorescence staining for Bcl-2 of heart sections from control and infected mice. Arrows and arrowheads indicate infiltrating and myocyte cells, respectively. Slides were observed and photographed using a Zeiss LSM Pascal confocal scanning microscope. Six animals per group were analyzed at different dpi. The results are expressed as mean ± SEM of all samples analyzed, with each P value being determined versus Control. Scale bar = 20 μm
Discussion
In the present study, we explored the functional relation between innate recognition of T. cruzi by cardiac cell primary cultures and the anti-apoptotic responses coordinated by the TLR2/IL6-dependent pathways. Herein, it was demonstrated that the triggering of TLR2 signaling, followed by IL6 production, plays an important role in the cardiomyocyte protection elicited by this parasite.
Although the dominant feature of the innate immune system is to protect the host from infectious agents, it may have other roles in mammalian biology. Thus, TLRs have been shown to be involved in tissue repair and homeostasis [20]. It has also been reported that TLR4 and TLR2 signaling confer a cardioprotective effect in both the isolated heart and/or cultured cardiomyocytes [14]. Interestingly, by transfection assays with a dominant negative form of TLR2 as well as by cultures obtained from C57BL/6 TLR2-knockout mice, we demonstrated that this receptor mediates the T. cruzi-induced anti-apoptotic effect. In agreement with this finding, it has been reported that TLR2 mediates anti-apoptotic effects and pro-inflammatory pathways in neonatal rat cardiomyocytes subjected to hydrogen peroxide [21]. In contrast, another study indicated that a TLR2 ligand induced apoptosis in HEK293 stably transfected with TLR2, but not in HEK293 cells which did not express TLR2, suggesting that this receptor also mediates pro-apoptotic signals [22]. The reason for this discrepancy is unclear but is probably multifactorial. Thus, the final outcome of TLR2 activation with respect to cell survival may well depend on the cell type (cell lines vs. primary cultures) and on the apoptotic models used.
In relation to our model, Petersen and colleagues demonstrated that TLR2 and NF-kB activation led to the production of IL1β, which mediated the cardiomyocyte hypertrophy elicited by T. cruzi [23]. In a later study, in agreement with our previous results, these authors found that T. cruzi prevented cell apoptosis through the activation of NF-kB, using neonatal rat cardiomyocytes subjected to serum starvation [24]. However, in contrast with our present findings, they found that the survival response was independent of TLR2 signaling in the T. cruzi-infected embryonic cardiac myoblast cell line H9c2, when tested by the lack of effect of TLR2 neutralizing antibodies. Nevertheless, it is not surprising that transformed myoblasts could not respond similarly to end-differentiated cardiac cells, as T. cruzi provides one of the most striking examples of how the outcome of infection is regulated in a cell type-specific manner [8, 10, 25–27].
Despite many studies having been carried out on IL6-related cytokines, clear physiological functions for IL6 in the stressed heart remain elusive. However, IL6 has roles that are consistent with the functional recovery of heart tissue following stress. Strikingly, IL6 exerted potent anti-apoptotic effects, affording complete protection of rat neonatal cardiomyocytes from sphingosine-induced death [2]. It has also been demonstrated that in vivo administration of IL6 completely reversed cardiac dysfunction and abrogated cardiomyocyte apoptosis in an experimental model of hypovolemic circulatory collapse [28]. These IL6 survival properties were mediated, at least in part, through activation of STAT3. Furthermore, during viral myocarditis, STAT3 phosphorylation has been shown to be involved in the cytoprotection of infected cells [29]. In agreement with these results, we found that IL6 released by cardiac cells in response to parasite infection fosters cellular survival and activates the STAT3 pathway. Although the mechanism remains to be elucidated, it is reasonable to speculate that it is through the gp130-STAT3 signaling pathway that IL6 conferred the cytoprotection observed in the present study. In the context of T. cruzi infection, the production and release of survival factors such as the pleiotropic cytokine IL6 could serve to block apoptosis in cells during the inflammatory process, thereby keeping them alive in a very unfavorable environment.
Regarding parasite components, it was reported that T. cruzi displays several ligands for TLRs. In particular, it was found that the tripomastigote-derived glycosylphosphatidylinositol (GPI) anchors linked to the surface mucin-like glycoproteins and free GPI anchors named glycoinositolphospholipids were recognized through TLR2 [16]. Additionally, T. cruzi Tc52-released protein induces human dendritic cell maturation signaling through TLR2 [30]. Thus, it is plausible to think that these parasite molecules might activate cardiomyocytes via TLR2, accounting for the protective responses elicited by tripomastigotes. Nevertheless, it is important to point out that living parasites may activate additional innate host responses through different TLRs other than TLR2 and/or TLR-independent mechanisms, in order to maintain the cardiac cell alive. Other studies performed with macrophages and T. cruzi infection have revealed that TLR2 is important for the induction of cytokine synthesis [16, 30], parasite internalization and replication [31]. In this cell type, the activation of TLR2 leads to the secretion of chemokines that regulate plasma extravasation into infected tissues, thus providing the substrates for the proteolytic generation of kinins by cruzipain [32]. Moreover, we have recently reported the crucial role of TLR2 and TLR9 in the control of the parasite replication and liver injury [33, 34].
The heart muscle, initially thought to be a bystander for T. cruzi infection, has been found to be an active participant in the immune response. Cardiac cell exposure to pro-inflammatory cytokines may precondition the myocardium environment to protect cardiomyocytes from apoptosis [35]. This hypothesis may be clinically supported by investigations performed on Chagasic patients with severe heart failure. Metzger et al. found that apoptotic cells were detected in high numbers in all samples examined. However, no apoptotic cardiomyocytes could be detected, and the majority of TUNEL-positive cells stained positively for the human macrophage marker CD68 [36], with similar results also being observed by other researchers [37] (see review by Higuchi et al. [38]). Related to this, we found an intense induction of Bcl-2 in cardiac fibers during the acute phase of the experimental infection, likely establishing a higher threshold against apoptosis in this cell type. In situ data obtained by immunofluorescence assays showed that the up-regulation of Bcl-2 occurred in both nonparasitized as well as in T. cruzi-parasitized cells in the inflamed cardiac tissue, reinforcing our in vitro observations and supporting the hypothesis that soluble factors secreted or released from the parasite (TLR2 ligands) or from the infected cells (IL6) could be responsible for cellular survival.
In conclusion, even though the exact mechanisms through which T. cruzi prevents cardiomyocyte apoptosis remain unknown, we have clearly shown a cross-talk between the intrinsic innate immune response of cardiomyocytes and the pro-survival effect evoked by the parasite.
Abbreviations
- mAchR:
-
M2 muscarinic acetylcholine receptor
- TPCK:
-
N-Tonsyl-l-phenylalanine chloromethyl ketone
- dnTLR2:
-
Dominant-negative TLR2
References
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388(6640):394–397
Craig R, Larkin A, Mingo AM, Thuerauf DJ, Andrews C, McDonough PM, Glembotski CC (2000) p38 MAPK and NF-kappa B collaborate to induce interleukin-6 gene expression and release. Evidence for a cytoprotective autocrine signaling pathway in a cardiac myocyte model system. J Biol Chem 275(31):23814–23824
Fredj S, Bescond J, Louault C, Delwail A, Lecron JC, Potreau D (2005) Role of interleukin-6 in cardiomyocyte/cardiac fibroblast interactions during myocyte hypertrophy and fibroblast proliferation. J Cell Physiol 204(2):428–436
Gao W, Pereira MA (2002) Interleukin-6 is required for parasite specific response and host resistance to Trypanosoma cruzi. Int J Parasitol 32(2):167–170
Savino W, Villa-Verde DM, Mendes-da-Cruz DA, Silva-Monteiro E, Perez AR, Aoki Mdel P, Bottasso O, Guinazu N, Silva-Barbosa SD, Gea S (2007) Cytokines and cell adhesion receptors in the regulation of immunity to Trypanosoma cruzi. Cytokine Growth Factor Rev 18(1–2):107–124
Gutierrez FR, Guedes PM, Gazzinelli RT, Silva JS (2009) The role of parasite persistence in pathogenesis of Chagas heart disease. Parasite Immunol 31(11):673–685. doi:10.1111/j.1365-3024.2009.01108.x
Brener Z (1973) Biology of Trypanosoma cruzi. Annu Rev Microbiol 27:347–382
Hashimoto M, Nakajima-Shimada J, Aoki T (2005) Trypanosoma cruzi posttranscriptionally up-regulates and exploits cellular FLIP for inhibition of death-inducing signal. Mol Biol Cell 16(8):3521–3528
Murata E, Hashimoto M, Aoki T (2008) Interaction between cFLIP and Itch, a ubiquitin ligase, is obstructed in Trypanosoma cruzi-infected human cells. Microbiol Immunol 52(11):539–543
Moore-Lai D, Rowland E (2004) Microarray data demonstrate that Trypanosoma cruzi downregulates the expression of apoptotic genes in BALB/c fibroblasts. J Parasitol 90(4):893–895
Chuenkova MV, PereiraPerrin M (2009) Trypanosoma cruzi targets Akt in host cells as an intracellular antiapoptotic strategy. Sci Signal 2(97):ra74. doi:10.1126/scisignal.2000374
Aoki MP, Guinazu NL, Pellegrini AV, Gotoh T, Masih DT, Gea S (2004) Cruzipain, a major Trypanosoma cruzi antigen, promotes arginase-2 expression and survival of neonatal mouse cardiomyocytes. Am J Physiol Cell Physiol 286(2):C206–C212
Aoki MP, Cano RC, Pellegrini AV, Tanos T, Guinazu NL, Coso OA, Gea S (2006) Different signaling pathways are involved in cardiomyocyte survival induced by a Trypanosoma cruzi glycoprotein. Microbes Infect 8(7):1723–1731. doi:S1286-4579(06)00117-1
Chao W (2009) Toll-like receptor signaling: a critical modulator of cell survival and ischemic injury in the heart. Am J Physiol Heart Circ Physiol 296(1):H1–H12
Ha T, Hu Y, Liu L, Lu C, McMullen JR, Kelley J, Kao RL, Williams DL, Gao X, Li C (2010) TLR2 ligands induce cardioprotection against ischaemia/reperfusion injury through a PI3 K/Akt-dependent mechanism. Cardiovasc Res 87(4):694–703. doi:10.1093/cvr/cvq116
Campos MAS, Almeida IC, Takeuchi O, Akira S, Valente EP, Procopio DO, Travassos LR, Smith JA, Golenbock DT, Gazzinelli RT (2001) Activation of toll-like receptor-2 by Glycosylphosphatidylinositol anchors from a protozoan parasite. J Immunol 167(1):416–423
Heussler VT, Machado J Jr, Fernandez PC, Botteron C, Chen CG, Pearse MJ, Dobbelaere DA (1999) The intracellular parasite Theileria parva protects infected T cells from apoptosis. Proc Natl Acad Sci USA 96(13):7312–7317
Fischer P, Hilfiker-Kleiner D (2007) Survival pathways in hypertrophy and heart failure: the gp130-STAT3 axis. Basic Res Cardiol 102(4):279–297. doi:10.1007/s00395-007-0658-z
Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R (2008) The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther 120(2):172–185
Medzhitov R (2010) Innate immunity: quo vadis? Nat Immunol 11 (7):551–553. doi:10.1038/ni0710-551
Frantz S, Kelly RA, Bourcier T (2001) Role of TLR-2 in the activation of nuclear factor kappaB by oxidative stress in cardiac myocytes. J Biol Chem 276(7):5197–5203
Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A (1999) Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285(5428):736–739
Petersen CA, Krumholz KA, Burleigh BA (2005) Toll-like receptor 2 regulates interleukin-1beta-dependent cardiomyocyte hypertrophy triggered by Trypanosoma cruzi. Infect Immun 73(10):6974–6980
Petersen CA, Krumholz KA, Carmen J, Sinai AP, Burleigh BA (2006) Trypanosoma cruzi infection and nuclear factor kappa B activation prevent apoptosis in cardiac cells. Infect Immun 74(3):1580–1587
Lopes MF, da Veiga VF, Santos AR, Fonseca ME, DosReis GA (1995) Activation-induced CD4 + T cell death by apoptosis in experimental Chagas ‘disease. J Immunol 154(2):744–752
Chuenkova MV, Furnari FB, Cavenee WK, Pereira MA (2001) Trypanosoma cruzi trans-sialidase: a potent and specific survival factor for human Schwann cells by means of phosphatidylinositol 3-kinase/Akt signaling. Proc Natl Acad Sci USA 98(17):9936–9941
Nakajima-Shimada J, Zou C, Takagi M, Umeda M, Nara T, Aoki T (2000) Inhibition of Fas-mediated apoptosis by Trypanosoma cruzi infection. Biochim Biophys Acta 1475(2):175–183
Alten JA, Moran A, Tsimelzon AI, Mastrangelo MA, Hilsenbeck SG, Poli V, Tweardy DJ (2008) Prevention of hypovolemic circulatory collapse by IL-6 activated Stat3. PLoS One 3(2):e1605. doi:10.1371/journal.pone.0001605
Yajima T, Yasukawa H, Jeon ES, Xiong D, Dorner A, Iwatate M, Nara M, Zhou H, Summers-Torres D, Hoshijima M, Chien KR, Yoshimura A, Knowlton KU (2006) Innate defense mechanism against virus infection within the cardiac myocyte requiring gp130-STAT3 signaling. Circulation 114(22):2364–2373. doi:10.1161/CIRCULATIONAHA.106.642454
Ouaissi A, Guilvard E, Delneste Y, Caron G, Magistrelli G, Herbault N, Thieblemont N, Jeannin P (2002) The Trypanosoma cruzi Tc52-released protein induces human dendritic cell maturation, signals via toll-like receptor 2, and confers protection against lethal infection. J Immunol 168(12):6366–6374
Maganto-Garcia E, Punzon C, Terhorst C, Fresno M (2008) Rab5 activation by Toll-like receptor 2 is required for Trypanosoma cruzi internalization and replication in macrophages. Traffic 9(8):1299–1315
Schmitz V, Svensjo E, Serra RR, Teixeira MM, Scharfstein J (2009) Proteolytic generation of kinins in tissues infected by Trypanosoma cruzi depends on CXC chemokine secretion by macrophages activated via Toll-like 2 receptors. J Leukoc Biol 85(6):1005–1014
Carrera-Silva EA, Cano RC, Guinazu NL, Aoki MP, Pellegrini AV, Gea S (2008) TLR2, TLR4 and TLR9 are differentially modulated in liver lethally injured from BALB/c and C57BL/6 mice during Trypanosoma cruzi acute infection. Mol Immunol 45(13):3580–3588. doi:10.1016/j.molimm.2008.05.004
Carrera-Silva EA, Guinazu N, Pellegrini A, Cano RC, Arocena A, Aoki MP, Gea S (2010) Importance of TLR2 on hepatic immune and non-immune cells to attenuate the strong inflammatory liver response during Trypanosoma cruzi acute infection. PLoS Negl Trop Dis 4(11):e863. doi:10.1371/journal.pntd.0000863
Smith RM, Lecour S, Sack MN (2002) Innate immunity and cardiac preconditioning: a putative intrinsic cardioprotective program. Cardiovasc Res 55(3):474–482
Metzger M, Higuchi ML, Brito T (2001) Lack of apoptosis of cardiomyocytes in chronic chagasic myocarditis. Virchow′s Archiv 439(6):352–359. doi:998
Rossi MA, Souza AC (1999) Is apoptosis a mechanism of cell death of cardiomyocytes in chronic chagasic myocarditis? Int J Cardiol 68(3):325–331. doi:10.1016/S0167-5273(98)00375-1
Higuchi ML, Benvenuti LA, Martins Reis M, Metzger M (2003) Pathophysiology of the heart in Chagas’ disease: current status and new developments. Cardiovasc Res 60(1):96–107. doi:10.1016/S0008-6363(03)00361-4
Acknowledgments
We are grateful to Dr. Joao Santana Silva (Universidade de Sao Paulo, Brazil) for the donation of the TLR2-knockout mice and for his help in the experiments performed in the Medical School of Ribeirão Preto, Department of Biochemistry and Immunology. We thank Alejandra Romero, Pilar Crespo, Paula Abadie, Cecilia Sampedro for their skillful technical assistance, and we would like to thank Dr. Paul Hobson, native speaker, for revision of the manuscript.
Ethics statement
All animal experiments were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health-USA. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Centro de Investigaciones en Bioquímica Clínica e Inmunología (CIBICI)-CONICET (Permit Number: 15-10-01).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ponce, N.E., Cano, R.C., Carrera-Silva, E.A. et al. Toll-like receptor-2 and interleukin-6 mediate cardiomyocyte protection from apoptosis during Trypanosoma cruzi murine infection. Med Microbiol Immunol 201, 145–155 (2012). https://doi.org/10.1007/s00430-011-0216-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-011-0216-z