
Abstract
Human immunodeficiency virus-1 tropism highly correlates with the amino acid (aa) composition of the third hypervariable region (V3) of gp120. A shift towards more positively charged aa is seen when binding to CXCR4 compared with CCR5 (X4 vs. R5 strains), especially positions 11 and 25 (11/25-rule) predicting X4 viruses in the presence of positively charged residues. At nucleotide levels, negatively or uncharged aa, e.g., aspartic and glutamic acid and glycine, which are encoded by the triplets GAN (guanine-adenosine-any nucleotide) or GGN are found more often in R5 strains. Positively charged aa such as arginine and lysine encoded by AAR or AGR (CGN) (R means A or G) are seen more frequently in X4 strains suggesting our hypothesis that a switch from R5 to X4 strains occurs via a G-to-A mutation. 1527 V3 sequences from three independent data sets of X4 and R5 strains were analysed with respect to their triplet composition. A higher number of G-containing triplets was found in R5 viruses, whereas X4 strains displayed a higher content of A-comprising triplets. These findings also support our hypothesis that G-to-A mutations are leading to the co-receptor switch from R5 to X4 strains. Causative agents for G-to-A mutations are the deaminases APOBEC3F and APOBEC3G. We therefore hypothesize that these proteins are one driving force facilitating the appearance of X4 variants. G-to-A mutations can lead to a switch from negatively to positively charged aa and a respective alteration of the net charge of gp120 resulting in a change of co-receptor usage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
The human immunodeficiency virus-1 (HIV) attaches to its target cells by binding its viral glycoprotein gp120 to the cellular receptor CD4. An additional co-receptor is required for successful cell entry. In general, HIV uses one of the chemokine receptors CXCR4 and CCR5 as co-receptor, in vivo [1]. The interaction between the co-receptor and gp120 is mediated mainly by V3 and by the co-receptor-binding site located in the bridging sheet of gp120 [2]. In more than 50% [3–6] of infected individuals, HIV entry requires a switch from CCR5 to CXCR4. This switch is associated with an accelerated decline of CD4+ and CD8+ T cells and a faster progression to the acquired immune deficiency syndrome (AIDS) in untreated patients [7–10].
With respect to protein biochemistry, the switch from CCR5 to CXCR4 is accompanied by an increase in the net charge of V3. This is often mediated by mutations from the negatively charged aspartic acid (Asp) and glutamic acid (Glu) to the positively charged arginine (Arg) and lysine (Lys) [11, 12]. Differences in the V3 amino acid composition are the basis of sequence-based predictions of the co-receptor usage [2, 13] (e.g. geno2pheno[coreceptor]). The 11/25 rule predicts a CXCR4-usage in case of a positive charge at position 11 and/or 25 [11, 14] and is commonly accepted due to its high specificity. In R5 viruses, these positions are filled by Asp, Glu, and Gly, encoded by the triplets GAN (guanine-adenosine-any nucleotide) and GGN. Interestingly, a single nucleotide mutation from G-to-A in the first position within these triplets can lead to the formation of the triplets AAR and AGR (R represents A or G), which encodes for the positively charged residues Lys and Arg predominantly found at the respective positions of X4 viruses.
Besides the viral reverse transcriptase (RT), which has an impact on the appearance of mutations, deletions and deletions with insertions within the HIV genome [15–17], APOBEC3F and APOBEC3G, which are members of the APOBEC3 (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide 3) protein family, could also be involved [18–21]. APOBEC3F and APOBEC3G have been identified as antiviral host factors of the innate immune system and their activity has been detected in the absence of HIV Vif [18–21]. APOBEC3G/F inhibit retroviral replication by deaminating cytidines (C) to uracil (U) in newly synthesized single-stranded DNA during reverse transcription. Consequently, G-to-A mutations can be observed in the viral genome [22–24]. While APOBEC3F preferentially mutates G into A in a GA-context, APOBEC3G prefers a GG-motif, resulting in a switch to AA and AG, respectively [18, 19, 21, 25]. It has been proposed that, in the absence of fully functional Vif, viral genomes are hypermutated to such an extent that they cannot produce infectious progeny viruses [22, 24, 26–28]. In contrast, cell culture experiments show that Vif does not cause a complete degradation of APOBEC3G: low-level catalytic activity of APOBEC3G was found [29] inducing mutations in HIV of long-term cultures [28]. This low-level activity of APOBEC3G might have a positive effect on viral infectivity by facilitating drug resistance [30], immune evasion [31, 32] and/or co-receptor switch as discussed herein.
After the identification of APOBEC3G, another member of this family with anti-HIV activity, APOBEC3F, was characterized. Both deaminases have substantial similarities, biochemically as well as functionally. However, most studies have been conducted on APOBEC3G, especially in T cells [33] or with recombinant APOBEC3G protein [34]. Both systems show that APOBEC3G exists in two forms, the high molecular mass (HMM) form as an inactive “precursor” and the low molecular mass (LMM) form having the antiretroviral activity. The presence of the LMM APOBEC3G in immature dendritic cells (DCs) might contribute to the low-level HIV replication observed in this cell type [35], in comparison with higher HIV replication in T cells [36]. Depending on the time point of activation, activating APOBEC3G with either a double-stranded RNA-homolog (poly(I: C)) or with type I Interferon could eradicate HIV entering the cells or synthesized de novo [37]. It has also been shown that APOBEC3G can be activated at transcriptional level by stimulating CCR5 [38]. Whether APOBEC3F does respond to the same stimuli needs to be determined.
We found in the V3 region of gp120, as the co-receptor important site, G-to-A mutations in a GA- and GG-context indicating an involvement of APOBEC3G/F. G-to-A mutations can lead to a switch from negatively to positively charged aa and a respective alteration of the net charge of gp120 resulting in a change of co-receptor usage. Therefore, we hypothesize that the APOBEC3G/F proteins are one but most likely not the only driving force facilitating the appearance of X4 variants.
Methods
Los Alamos data set
Nucleotide sequences with experimentally determined R5 or X4 co-receptor tropism were downloaded from the Los Alamos Sequence Database (db). To avoid bias from over-represented patients, at most one R5 and one X4 sequence per patient was randomly chosen. This resulted in a data set of 1026 V3 sequences (874 R5, 152 X4). Sequences were translated and aligned using CLUSTALW [39]. The alignment was done using the geno2pheno[coreceptor] alignment procedure (http://www.geno2pheno.org). For this, each aa sequence is individually aligned against a reference profile containing gap-columns at positions that typically display insertions. Based on these aa alignments, the nucleotide sequences were aligned. According to tropism, alignments were split into sets of R5 and X4 sequences. The percentage of the four nucleotides (A, C, T and G) at each position in V3 for each data set was calculated.
HIV-GRADE clinical data set
A total of 501 samples (349 R5, 152 X4) from patients screened for Maraviroc therapy were sequenced and collected by the HIV-GRADE consortium (http://www.hiv-grade.de), accession numbers HQ117318–HQ117818. In addition, the co-receptor usage of these samples was determined with the Monogram Trofile Tropism Assay. Data were analysed as described above. Numbering of both, Los Alamos and HIV-GRADE, data sets is according to the V3 sequence of consensus B (TGTACAAGACCCAACAACAATACAAGAAAAAGTATACATATAGGACCAGGGAGAGCATTTTATACAACAGGAGAAATAATAGGAGATATAAGACAAGCACATTGT, Los Alamos HIV Sequence Database, November 16th 2009).
PCR amplification of the HIV gp120 V3 region (longitudinal samples)
Reverse transcription polymerase chain reaction (RT–PCR) and nested PCR were used to amplify gp120 from plasma samples. The RT–PCR was performed using the OneStep RT–PCR System QIAamp OneStep RT–PCR kit (Qiagen, Hilden, Germany) according to the manufacture’s instructions. Using an external primer pair, V3F and V3R (Table 1) at the conditions listed in Tables 2 and 3, a PCR product of approximately 1.6 kb was generated. If required, a nested PCR was performed to generate a 0.8-kb product using the HotStarTaq kit (Qiagen) and the primer pair V3-1 and V3-5 (Table 1). The cycler conditions for the nested PCR are listed in Table 3. A PCR product of approximately 0.5 kb was generated.
Sequencing of the HIV gp120 V3 region
PCR products were used for multiple sequencing using BigDye TaqCycle sequencing kit v1.1 (Applied Biosystems, Darmstadt, Germany). Multiple primers (Table 1) were used to generate sequences. The conditions are listed in Table 4. Sequence analysis was acquired using the ABI 3130xl Genetic Analyzer (Applied Biosystems) and DNASTAR Lasergene SeqMan Pro (GATC Biotech AG, Konstanz, Germany). Sequences are published at GenBank accession numbers HQ117819–HQ117844.
Statistical analysis
The frequencies of the different nucleotides in V3 within R5 and X4 strains were correlated as follows: We first computed the frequencies fR5/X4,position(nt) of the nucleotides at the different positions within the R5 and X4 sets, e.g. fX4,4(G) displays the frequency of a G at position 4 within the X4 data set. We then calculated the difference dposition(nt) = fR5,position(nt) − fX4,position(nt) between the frequencies in R5 and X4 sequences, reflecting a possible shift from one nucleotide to another. Finally, we correlated the differences of one nucleotide with the differences of another nucleotide using Pearson’s product–moment correlation coefficient Rho (R), a measure of linear relationship. The rational behind this is that if we, e.g., often have a mutation from C to T at a given position (pos), then dpos(C) should roughly be −dpos(T), the relative decrease in C’s in R5 viruses should be compensated for by a similarly increased frequency of T’s. If there is no such directed mutation towards T, one also would not expect these numbers to lie in the same range.
Results
GAN or GGN triplets are found more often in V3 regions of R5 and AAR or AGR in V3 regions of X4 strains
The V3 net charge is one determinant for the prediction of HIV-1 co-receptor usage. While a lower net charge (≤+5) is associated with a tight gp120-CCR5 binding, a net charge of ≥+5 facilitates a gp120 interaction with CXCR4 [40, 41]. Since the amino acid composition plays a major role, we further analysed the nucleotide sequences in triplets, the basis of the genetic code (supplementary Table 1). V3 nucleotide sequences of classified R5 and X4 strains from 501 clinical samples (HIV-GRADE, Fig. 1, Table 5) as well as 1026 sequences originating from the Los Alamos HIV Sequence Database (supplementary Fig. 1, Table 5) were analysed for their triplet composition. By calculating the average triplet composition, we found in the HIV-GRADE data set a reduction of GAN triplets from 1.61 (R5) to 1.45 (X4) (Table 5). In contrast, AAR triplets occurred in R5 strains 1.06 times on average and 1.67 times in X4 strains (Table 5). The same pattern was observed for the sequences from the Los Alamos HIV Sequence Database. While the average GAN amount was reduced from 1.67 in R5 to 1.27 in X4, AAR triplets occurred in R5 strains 0.96 on average and in X4 strains 1.50 (Table 5). In addition, we observed in the HIV-GRADE data set a reduction of GGN triplets from 4.19 (R5) to 4.00 (X4) (Table 5). AGR triplets occurred in R5 strains on average of 3.81 and of 4.11 in X4 strains (Table 5). The same trend showing a reduction of GGN triplets from R5 to X4 as well as an enhancement of AGR triplets from R5 to X4 was observed for the data from the Los Alamos Database: while the average GGN amount was reduced from 4.18 in R5 to 4.07 in X4, the average number of AGR triplets was 3.72 in R5 strains and 4.13 in X4 strains (Table 5).

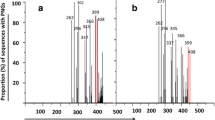
Triplet and amino acid (aa) distribution in R5 and X4 strains. V3 sequences from the HIV-GRADE data set were phenotyped with the Monogram Trofile Tropism Assay. According to the aa alignment, nucleotides were aligned and compared for their appearance in R5 (black) and X4 (grey). The percentage of each triplet (adenosine (abbreviated A), cytosine (C), guanine (G) and thymine (T) and the corresponding aa (Ala, alanine; Arg, arginine; Asn, aspargine; Asp, aspartic acid; Gln, glutamine; Glu, glutamic acid; Gly, glycine; Lys, lysine; Ser, serine; Thr, threonine, supplementary Table 1) for the position 11, 22, 24, and 25 are shown. Amino acids are grouped with respect to their physicochemical characteristics
In order to analyse the amino acid composition and their respective triplets, we compared the sequences of those two data sets, HIV-GRADE and Los Alamos HIV Sequence Database. We found four positions that differed strongly between R5 and X4 strains with respect to amino acid characteristics (e.g. charge) and triplet composition. Sequences of the HIV-GRADE data set (Fig. 1) showed at position 11 an AGT (adenosine-guanine-thymine, coding for serine, Ser) in 68.2% of the R5 strains in contrast to 42.8% of the X4 strains. The second most frequent triplet was GGT (Gly) appearing in 22.9% of the R5 and in 26.3% of the X4 sequences. In addition, we found at position 11 a CGT (Arg) in 2.6% of the X4 sequences, an AGA (Arg) in 5.3%, and an AGG (Arg) in 8.6%, all encoding the positively charged arginine (Arg). Furthermore, 2.6% of the X4 viruses harboured an AAG, encoding the positively charged amino acid lysine (Lys). For comparison: just 0.3% of the R5 strains had one of these triplets (namely AGG) at this location. With respect to position 22, 70.8% of the R5 strains had a GCA (guanine-cytosine-adenosine, alanine, Ala) and 23.2%, an ACA (threonine, Thr) compared with 50% with GCA and 40.1% with ACA (threonine, Thr) in X4 viruses. We noticed a deletion of the triplet at position 24 in 13.2% of the R5 and 17.8% of the X4 strains (data not shown). Considering only sequences without this deletion, we found that R5 mainly (81.2%) had a GGA (glycine, Gly), whereas X4 viruses were more diverse: 61.6% had a GGA (Gly), 12.8% a GAA (Glu), 5.6% an AAA (Lys), 4% a GAC, and 3.2% a GAT (both aspartic acid, Asp).
At position 25 of the V3 region of R5 strains, 12.9% had a GAT and 33.5% a GAC encoding for the negatively charged Asp, 3.4% had GAG and 23.2% a GAA encoding for glutamic acid (Glu), another negatively charged aa. Additionally, 3.2% had an AAC (asparagine, Asn) and 8.6% a CAA (glutamine, Gln) giving the site a neutral character. In contrast, the V3 region of X4 viruses showed only 5.3% GAT and 19.7% GAC (Asp) at this position as well as 2% GAG and 15.1% GAA (Glu). Furthermore, 5.9% AAC (Asn) and 6% CAA (Gln) were found. Uniquely for X4, 8.6% AGA (Arg) and 15.1% AAA (Lys) were identified. Both Arg and Lys are positively charged and contribute to the positive V3 net charge of X4 strains reflected by the 11/25-rule. The data collected from the Los Alamos HIV Sequence Database gave similar results (supplementary Fig. 1). Taken together, GAN and GGN triplets are found more often in V3 regions of R5 and AAR and AGR in X4 strains. Furthermore, in agreement with already published data [11, 14], these results show a difference between R5 and X4 strains at positions 11, 22, 24, and 25.
Differences in R5 and X4 strains occur mainly through G-to-A mutations
By looking at the nucleotide level, we noticed that R5 strains have more G-containing and X4 strains have more A-containing triplets, especially at positions 11, 22, 24 and 25. To confirm our hypothesis, we analysed the V3 sequences with respect to the possibility that the APOBEC3 proteins might be responsible for these differences. APOBEC3F favours GA as recognition sequence and APOBEC3G prefers a GG-motif resulting in a mutation to AA and AG, respectively (Fig. 2a). In the triplets at positions 22, 24 and 25, changes from G to A were found (Fig. 2b) in at least half of the sequences. In detail, GCA (Ala) was the predominant triplet at position 22 in R5 strains, whereas in X4 strains mainly ACA (Thr) was found. Furthermore, for position 24, GGA (Gly) was identified as the main triplet in R5 strains, whereas in X4 strains GAA (Glu), AAA (Lys), AGA (Arg), ACA (Thr),and GAC (Asp) were observed. AGA, AAA and GAA fulfil the requirements of a G-to-A mutation; ACA and GAC must have evolved through different mechanisms. At position 25, the main triplets in R5 strains are GAT, GAC as well as GAA. In contrast, X4 strains showed AAT, AAC, AAA, AGA and CAA. The mutation of the G in the triplets GAT, GAC and GAA (R5) resulted in the corresponding triplets AAT, AAC and AAA, but not directly in AGA or CAA in X4. However, position 11 showed AGT (R5) and AGG, AGA, CGT, and AAG (X4) which did not follow the expected pattern indicating that other factors such as the RT plays also a major role in nucleotide substitution [15–17]. In order to identify the deaminase, which might be responsible for the G-to-A mutations, a closer look was taken at the context in which they appeared (Fig. 2a). It seems that in triplet 24 and 25, both APOBEC3G and APOBEC3F, are the main force causing the switch from R5 to X4 viruses. Triplet 24 GGA in R5 showed both of the preferred motifs (GG and GA) of the two deaminases. Furthermore, all possible combinations of triplets (AGA, GAA and AAA) resulting from G-to-A mutations for this triplet were found in X4 viruses. In R5 strains, position 25 contained mainly the preferred motif of APOBEC3F coding for the corresponding mutation in X4 viruses. These findings suggest, in addition to the fidelity of the RT, a mechanism based on the cytidine deaminase function of APOBEC3G/F. The activity of APOBEC3G/F results in a G-to-A mutation, but not always in a hypermutation. However, APOBEC3G and F do not seem to be exclusively responsible for the co-receptor switch as seen at position 11.

Summary of triplets and the respective amino acids divided in R5 and X4 HIV. a Preferred recognition motifs (adenosine (abbreviated A), guanine (G)) and resulting mutations after deamination of APOBEC3F (bold) and APOBEC3G (boxed). b Triplets at positions 11, 22, 24 and 25 in R5 (left) and X4 (right) are depicted (adenosine (abbreviated A), cytosine (C), guanine (G) and thymine (T). Arrows represent the changes from R5 to X4 in case of a G-to-A transition, whereas arrows with dashed lines indicate G-to-A independent changes. G-to-A changes that might be caused by APOBEC3F are bold and changes caused by APOBEC3G are boxed. Amino acids (abbreviation as in Fig. 1, supplementary Table 1) are grouped with respect position and physicochemical characteristics are declared
In R5 versus X4 strains G-to-A mutations correlate at distinctive amino acid positions
The HIV-GRADE and the Los Alamos data set are cross-sectional data sets. Therefore, it is not possible to differentiate whether a nucleotide appeared through a mutation from another or whether it was always there. Another problem is that in contrast to the polymerase, the envelope protein gp120 is due to immune selection highly polymorphic, especially within the variable loops. Therefore, there are no conserved wild-type sequences, which could be used as a template and against which one could compare these sequences. Hence, mutations from one residue to another can only be inferred indirectly. We accounted for this by correlating differences in nucleotide frequencies between R5 and X4 strains as described in the method section. We separately analysed the two independent data sets (HIV-GRADE (Fig. 3a, supplementary Fig. 2A) and Los Alamos (Fig. 3b, supplementary Fig. 2B)) and graphed the correlations of each nucleotide pair, namely d(G) versus d(C), d(G) versus d(T), d(A) versus d(T), d(C) versus d(A), d(C) versus d(T), and d(G) versus d(A) (Fig. 3a/b, supplementary Fig. 2A/B). The largest R-value resulted in the differences (d) of G’s and A’s (d(G) versus d(A)) with a value of −0.71 (Fig. 3a). The analysis of the data set from the Los Alamos HIV Sequence Database (Fig. 3b, supplementary Fig. 2B) showed similar results. Again, the largest R-value was calculated for the correlation of d(G) versus d(A) (R = −0.53) (Fig. 3b). All other correlations were much weaker (R values of at most −0.36, supplementary Fig. 2A/B). The second and fourth quadrants (upper left and lower right) of Fig. 3 and supplementary Fig. 2A/B describe positions with reverse differences, i.e. where one nucleotide was more often seen in R5 strains while the other nucleotide was seen more in X4 viruses, or vice verse, e.g. in case of the G–A correlation, a decrease of G’s in R5 strains associated with an increase of A’s in X4 strains is found in the fourth quadrant. The higher the difference between the frequencies in the two phenotypes, the higher is the distance from zero. To highlight these positions, we enlarged the fourth quadrant of the G–A correlation in both data sets showing the area of x above 0.05 and y below −0.03 (Fig. 3a/b, nucleotide (nt) position/triplet (tp) position). Within the HIV-GRADE data set (Fig. 3a), we found the most significant changes in G-to-A mutations at positions nt26/tp9, 32/11, 64/22, 70/24, 71/24 and 73/25. The correlations from the V3 sequences of the Los Alamos HIV Sequence Database occur at the same positions, namely 32/11, 64/22, 70/24, 71/24 and 73/25 (Fig. 3b). In summary, both cross-sectional data sets, HIV-GRADE as well as Los Alamos, have more G-containing triplets in R5 than in X4 viruses at key positions important for co-receptor usage, while the latter have at these positions more A-containing triplets than R5 strains. This suggests the involvement of a member of the APOBEC3 family, such as APOBEC3G/F as one driving force facilitating the switch from CCR5- to CXCR4-usage of HIV.

G-to-A changes from R5 to X4 strains show highest correlation. The differences at each position in V3 between R5 and X4 strains regarding the percentage of nucleotide pairs (adenosine (abbreviated A) and guanine (G) from the HIV-GRADE (a) and the Los Alamos (b) data set were calculated and correlated. Based on this calculation, the positions with the highest differences in R5 versus X4 that mutate from one nucleotide to the other are located in the fourth (lower right) quadrant. The greatest correlation is boxed in red and enlarged (x ≥ 0.05, y ≤ −0.03). Nucleotide and triplet positions are indicated and separated by a slash
Longitudinal data of ten patients confirm cross-sectional data
Based on the analysis of cross-sectional data, we hypothesize that the co-receptor switch is mediated by APOBEC3F and APOBEC3G, at least in part, since G-to-A transitions occur more often than others, especially on sites important for co-receptor usage. To further test this hypothesis, 26 longitudinal V3 sequences from ten patients (Fig. 4) were analysed. The time interval between samples was at least 1 month and the co-receptor usage was predicted using geno2pheno[coreceptor] with a false positive rate (fpr) below 20% considered as predictive for CXCR4-usage. Only the aa at position 11, 22, 23, 24 and 25 are shown (Fig. 4) because only these displayed G-to-A mutations in the longitudinal samples. G-to-A mutations at positions 22, 24 and 25 are in agreement with the Los Alamos and HIV-GRADE data sets. As highlighted in bolded letters within the nucleotide alignment, G-to-A mutations occurred in longitudinal samples and seem to be part of the mechanism for the co-receptor switch (Fig. 4). We found both, the recognition motif GG for APOBEC3G (light grey), e.g. in patient (pat.) 1, triplet 11, or in pat. 4, triplet 24, and the recognition motif GA for APOBEC3F (black), e.g. pat. 4 and 6, triplets 25. In our 10 patients, we found five APOBEC3G-based (pat. 1, triplet 11; pat. 4, triplet 24; pat. 5, triplet 11; pat. 8, triplet 24/25, pat. 10, triplet 11) and five APOBEC3F-based (pat. 3, triplet 25; pat. 4, triplet 25; pat. 5, triplet 22/23; pat. 6, triplet 25; pat. 10, triplet 24 and 25) mutations. In addition, we also found seven G-to-A transitions independent of the two motifs, GG and GA. However, three of those (framed) display one of the recognition motifs not following the expected pathway. In particular, pat. 1 showed in triplet 25, the APOBEC3G recognition motif GG, which changed in the next sample to the resulting APOBEC3F sequence AA (instead of AG). In pat. 3, we found in triplet 23 a mixed population containing, in addition to the APOBEC3G recognition motif GG, also a G/C sequence. The analysis of the next viral sequence from this patient 4 months later displayed an A/C at this position. We found also in pat. 5, triplet 11, month 0 a mixed viral population containing the APOBEC3G recognition motif GG and the corresponding mutation AG (light grey). We also found for pat. 5 in triplet 23 and 24 (nts 69 and 70), a mixed population containing possibly both recognition motifs (framed). While after 12 months an A became the dominant nucleotide (nt 69), the second triplet (triplet 24) was deleted (—). Furthermore, triplet 24 showed deletions (—) (pat. 1, month 38; pat. 2, month 4; pat. 5, month 12) and an insertion (pat. 3, month >4), which of course could also have an impact on changing the tropism. The appearance of deletions is not caused by the APOBEC-proteins but most likely by the activity of the RT [16]. In total, 9 of the 10 patients showed G-to-A mutations possibly leading to or influencing co-receptor switch (pat. 2 shows only a deletion in triplet 24). Besides nucleotide changes noticed in the recognition motifs of APOBEC3G and APOBEC3F, we also found APOBEC3G/F independent G-to-A mutations being one mechanism responsible for the co-receptor switch of HIV-1.

Potential involvement of APOBEC3G and APOBEC3F in G-to-A changes in longitudinal samples of ten patients. Nucleotide alignment of ten patients (pat. 1–10) showing a switch in co-receptor usage or a drop of the false positive rate (fpr). The fpr < 20 is representing X4 and highlighted in bolded numbers. Shown are triplets 11, 22, 23, 24, and 25 (adenosine (abbreviated A), cytosine (C), guanine (G), thymine (T), deletion (–)) and the corresponding nucleotide positions per column. All detected nucleotides of mixed populations are shown and divided by a slash, whereas the major population is indicated by the capital letter. The APOBEC3G recognition motif (GG) with a subsequent G-to-A mutation is highlighted in light grey. The APOBEC3F recognition motif (GA) with a G-to-A change is marked in black. G-to-A changes that appear independent of an APOBEC-related recognition motif are bolded. Sequences with one of the recognition motifs that do not follow the expected pathway are framed
Discussion
Here, we describe an increase of G-to-A mutations comparing R5 and X4 HIV on both, cross-sectional data as well as longitudinal data on sites within the V3 region important for co-receptor usage. Data were obtained from three independent sources: HIV-GRADE, Los Alamos HIV Sequence Database, and a collection of longitudinal patient samples evaluated at the Institute of Virology, Cologne, Germany. By analysing these three sets, we found in co-receptor relevant sites more GAN triplets in R5 and more AAR triplets in X4 strains (Fig. 1, supplementary Fig. 1 and Fig. 2B), suggesting an APOBEC3 cytidine deaminase-mediated co-receptor switch from CCR5 to CXCR4.
The correlation data of both, HIV-GRADE and Los Alamos, showed more G-to-A mutations than every other substitution. Most of the G-to-A mutations appeared at positions 22, 24 and 25, especially positions 24 and 25 are known to be important for co-receptor usage [11, 14] implying that APOBEC proteins could play a role in the evolution towards X4 strains. In particular, position 25 showed in R5 the triplet GAA and in X4 the triplet AAA supporting our hypothesis. In contrast, position 11, also very important for co-receptor usage, did not show such an obvious pattern in most sequences. At position 11 we found in R5 strains mainly AGT. A G-to-A mutation would lead to an AAT (Fig. 2B). Nevertheless, this triplet was not found at this position, neither in R5 nor in X4 strains (Fig. 1, supplementary Fig. 1). This suggests that the asparagine residue at this position is disadvantageous for the virus. In addition to G-to-A mutations, C-to-T mutations were also seen frequently. They could be the consequence of the deamination of methylated cytosine residues within the integrated HIV genome resulting in thymidine instead [42].
Two candidates of the APOBEC3 family, namely APOBEC3G and APOBEC3F, seem to be involved. Our data support a slightly higher activity of APOBEC3F because more deamination was seen in the GA context (Figs. 2, 4). However, the infidelity of the RT might have a substantial impact on these nucleotide substitutions [15–17]. As described by Preston et al., the preference of nucleotide mismatches of A:C > A:G > A:A [17] with a high preference of G-to-A mutations (frequency 7 × 10−6 per base pair per cycle) [15] and a deletion rate with a frequency of 2 × 10−6 [16] must not be underestimated. However, the summary of the HIV-GRADE and Los Alamos data showed the APOBEC3G recognition motif only at position 24, whereas the APOBEC3F recognition pattern with the resulting mutations were seen in both, position 24 and 25 (Fig. 2). The work of Han et al. who compared the activity of APOBEC3G and APOBEC3F showed in CEM-SS T cells that APOBEC3G inhibits viral replication of Vif-deficient HIV stronger than APOBEC3F [43]. This suggests that APOBEC3G causes more often hypermutated, replication incompetent viruses [22, 24, 26–28], whereas APOBEC3F activity resulted more often in mutated but replicating viruses. One possible mechanism is suitable for APOBEC3G, which can change TGG (Trp) in TAG (Stop) leading to a strong inhibition of viral replication. However, both APOBEC3 proteins cause mutations leading to drug resistance [30], immune evasion [31, 32] and/or possibly co-receptor switch as we suggest here. The analysis of the 26 longitudinal samples from ten individuals resulted in five APOBEC3G- and seven APOBEC3F-based G-to-A mutations (Fig. 4). Two samples from pat. 4 were available. While the first sequence was predicted as R5 (fpr 60.8%) the second, which was collected 41 months later, was predicted as X4 (1.7%). The comparison of the two sequences clearly shows on APOBEC3G-based mutation in triplet 24 and an APOBEC3F-based mutation in triplet 25. We also detected mixed populations (e.g. pat. 5, triplet 11, GGT/AGT, Gly/Ser) representing the existence of two major virus populations. One could speculate that the population with the AGT genotype might have been evolved from the GGT genotype after the deamination activity of APOBEC3G. Furthermore, APOBEC3G/F-independent mutations were found. They might have been evolved either non-specifically by APOBEC3G/F or by other mechanisms such as the RT fidelity [3–6, 15–17]. This is shown in pat. 9 where the fpr drops from 90% down to 1.7% 23 months later only showing G-to-A mutations in triplet 11 and 22 without an APOBEC3-recognition motif.
In summary, our data from three independent sources, HIV-GRADE, Los Alamos and longitudinal data sets, strongly suggest an involvement of APOBEC3 deaminases in the background of the RT fidelity. Their activity in deaminating cytidines resulting in a change from G to A in the viral template with the effect that the aa composition of the V3 region is changed in a way that co-receptor usage switches from CCR5 to CXCR4. Our findings are leading to the question: Does HIV exploit APOBEC proteins to its own benefit? Mulder et al. [30] demonstrated that APOBEC3G facilitates drug-resistance mutations in HIV in the presence of 3TC in vitro. Protease inhibitor resistance mutations were observed most frequently when APOBEC3G and APOBEC3F dinucleotide target motifs were mutated [31]. Furthermore, due to the APOBEC-mediated effect, CTL-specific epitopes within the viral protease [31] and the envelope proteins [32] are mutated leading to immune evasion. With respect to co-receptor switch, Pido-Lopez et al. [38] have shown that stimulating the CCR5 receptor with its natural ligand CCL3 (Mip-1α) increases the mRNA levels of APOBEC3G. It is possible that by binding its co-receptor R5 HIV induces the expression of APOBEC proteins and thereby facilitates mutations in the co-receptor relevant sites.
Conclusions
Our data support the hypothesis that the antiviral potential of APOBEC3G/F and the RT might also be used by the virus to create genetic diversity in order to adapt to the host-specific environment in the presence and absence of antiviral drugs. In particular, both, APOBEC3G and APOBEC3F seem to facilitate co-receptor switch by mutating co-receptor relevant sites within the V3 region of gp120. This mechanism needs to be further explored in vitro as well as in vivo. Additionally, it would be useful to control for this mechanism by using vif-deficient virus when testing the development of drug resistance mutations in vitro. Furthermore, it could be possible that the use of CCR5 antagonists, such as Maraviroc, inhibits the expression of APOBEC3G/F by blocking the CCR5 signalling. Therefore, our data open further perspectives of the role of APOBEC proteins in the HIV infection and disease progression.
References
Berger EA, Murphy PM, Farber JM (1999) Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol 17:657–700
Sierra S, Kaiser R, Thielen A, Lengauer T (2007) Genotypic coreceptor analysis. Eur J Med Res 12(9):453–462
Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI (1999) Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol 73(12):10489–10502
Schuitemaker H, Koot M, Kootstra NA, Dercksen MW, de Goede RE, van Steenwijk RP, Lange JM, Schattenkerk JK, Miedema F, Tersmette M (1992) Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J Virol 66(3):1354–1360
Moyle GJ, Wildfire A, Mandalia S, Mayer H, Goodrich J, Whitcomb J, Gazzard BG (2005) Epidemiology and predictive factors for chemokine receptor use in HIV-1 infection. J Infect Dis 191(6):866–872. doi:10.1086/428096
Bozzette SA, McCutchan JA, Spector SA, Wright B, Richman DD (1993) A cross-sectional comparison of persons with syncytium- and non-syncytium-inducing human immunodeficiency virus. J Infect Dis 168(6):1374–1379
Alimonti JB, Ball TB, Fowke KR (2003) Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. J Gen Virol 84(Pt 7):1649–1661
Rosenberg YJ, Anderson AO, Pabst R (1998) HIV-induced decline in blood CD4/CD8 ratios: viral killing or altered lymphocyte trafficking? Immunol Today 19(1):10–17
Herbein G, Mahlknecht U, Batliwalla F, Gregersen P, Pappas T, Butler J, O’Brien WA, Verdin E (1998) Apoptosis of CD8+ T cells is mediated by macrophages through interaction of HIV gp120 with chemokine receptor CXCR4. Nature 395(6698):189–194
Zhang SY, Zhang Z, Fu JL, Kang FB, Xu XS, Nie WM, Zhou CB, Zhao M, Wang FS (2009) Progressive CD127 down-regulation correlates with increased apoptosis of CD8 T cells during chronic HIV-1 infection. Eur J Immunol 39(5):1425–1434
Delobel P, Nugeyre MT, Cazabat M, Pasquier C, Marchou B, Massip P, Barre-Sinoussi F, Israel N, Izopet J (2007) Population-based sequencing of the V3 region of env for predicting the coreceptor usage of human immunodeficiency virus type 1 quasispecies. J Clin Microbiol 45(5):1572–1580
De Jong JJ, De Ronde A, Keulen W, Tersmette M, Goudsmit J (1992) Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J Virol 66(11):6777–6780
Lengauer T, Sander O, Sierra S, Thielen A, Kaiser R (2007) Bioinformatics prediction of HIV coreceptor usage. Nat Biotechnol 25(12):1407–1410
Raymond S, Delobel P, Mavigner M, Cazabat M, Souyris C, Sandres-Saune K, Cuzin L, Marchou B, Massip P, Izopet J (2008) Correlation between genotypic predictions based on V3 sequences and phenotypic determination of HIV-1 tropism. Aids 22(14):F11–F16
Pathak VK, Temin HM (1990) Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: substitutions, frameshifts, and hypermutations. Proc Natl Acad Sci USA 87(16):6019–6023
Pathak VK, Temin HM (1990) Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: deletions and deletions with insertions. Proc Natl Acad Sci USA 87(16):6024–6028
Preston BD, Poiesz BJ, Loeb LA (1988) Fidelity of HIV-1 reverse transcriptase. Science 242(4882):1168–1171
Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH (2004) Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol 14(15):1392–1396
Bishop KN, Holmes RK, Sheehy AM, Malim MH (2004) APOBEC-mediated editing of viral RNA. Science 305(5684):645
Sheehy AM, Gaddis NC, Choi JD, Malim MH (2002) Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418(6898):646–650
Wiegand HL, Doehle BP, Bogerd HP, Cullen BR (2004) A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J 23(12):2451–2458
Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH (2003) DNA deamination mediates innate immunity to retroviral infection. Cell 113(6):803–809
Lecossier D, Bouchonnet F, Clavel F, Hance AJ (2003) Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 300(5622):1112
Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D (2003) Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424(6944):99–103
Liddament MT, Brown WL, Schumacher AJ, Harris RS (2004) APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr Biol 14(15):1385–1391
Pace C, Keller J, Nolan D, James I, Gaudieri S, Moore C, Mallal S (2006) Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. J Virol 80(18):9259–9269
Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD (2005) Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog 1(1):e6
Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L (2003) The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424(6944):94–98
Nowarski R, Britan-Rosich E, Shiloach T, Kotler M (2008) Hypermutation by intersegmental transfer of APOBEC3G cytidine deaminase. Nat Struct Mol Biol 15(10):1059–1066
Mulder LC, Harari A, Simon V (2008) Cytidine deamination induced HIV-1 drug resistance. Proc Natl Acad Sci USA 105(14):5501–5506
Pillai SK, Wong JK, Barbour JD (2008) Turning up the volume on mutational pressure: is more of a good thing always better? (a case study of HIV-1 Vif and APOBEC3). Retrovirology 5:26
Wood N, Bhattacharya T, Keele BF, Giorgi E, Liu M, Gaschen B, Daniels M, Ferrari G, Haynes BF, McMichael A, Shaw GM, Hahn BH, Korber B, Seoighe C (2009) HIV evolution in early infection: selection pressures, patterns of insertion and deletion, and the impact of APOBEC. PLoS Pathog 5(5):e1000414
Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC (2005) Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature 435(7038):108–114
Chelico L, Pham P, Calabrese P, Goodman MF (2006) APOBEC3G DNA deaminase acts processively 3′–>5′ on single-stranded DNA. Nat Struct Mol Biol 13(5):392–399
Pion M, Granelli-Piperno A, Mangeat B, Stalder R, Correa R, Steinman RM, Piguet V (2006) APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J Exp Med 203(13):2887–2893
Kreisberg JF, Yonemoto W, Greene WC (2006) Endogenous factors enhance HIV infection of tissue naive CD4 T cells by stimulating high molecular mass APOBEC3G complex formation. J Exp Med 203(4):865–870
Trapp S, Derby NR, Singer R, Shaw A, Williams VG, Turville SG, Bess JW Jr, Lifson JD, Robbiani M (2009) Double-stranded RNA analog poly(I:C) inhibits human immunodeficiency virus amplification in dendritic cells via type I interferon-mediated activation of APOBEC3G. J Virol 83(2):884–895
Pido-Lopez J, Whittall T, Wang Y, Bergmeier LA, Babaahmady K, Singh M, Lehner T (2007) Stimulation of cell surface CCR5 and CD40 molecules by their ligands or by HSP70 up-regulates APOBEC3G expression in CD4(+) T cells and dendritic cells. J Immunol 178(3):1671–1679
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680
Fouchier RA, Groenink M, Kootstra NA, Tersmette M, Huisman HG, Miedema F, Schuitemaker H (1992) Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J Virol 66(5):3183–3187
Briggs DR, Tuttle DL, Sleasman JW, Goodenow MM (2000) Envelope V3 amino acid sequence predicts HIV-1 phenotype (co-receptor usage and tropism for macrophages). Aids 14(18):2937–2939
Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E (2009) Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog 5(6):e1000495
Han Y, Wang X, Dang Y, Zheng YH (2008) APOBEC3G and APOBEC3F require an endogenous cofactor to block HIV-1 replication. PLoS Pathog 4(7):e1000095
Acknowledgments
The authors would like to thank all members of the laboratory, especially Finja Schweitzer, for critical discussions throughout the study. The professional technical assistance of Dörte Hammerschmidt and Monika Timmen-Wego is gratefully acknowledged. Additional thanks goes to the HIV-GRADE consortium, especially to Mark Oette, Krankenhaus der Augustinerinnen, Cologne; Gerd Fätkenheuer, University Hospital of Cologne, Cologne; Stefan Esser, University of Duisburg-Essen, Essen; Stefan Reuter, University of Düsseldorf, Düsseldorf; all Germany for providing the clinical data set. This work was supported by German Bundesministerium für Gesundheit grant number 310/4476-02/3, the German Bundesministerium für Bildung und Forschung grant number 01ES0712 and grant number 0315480C, and the European Commission Health project Collaborative HIV and Anti-HIV Drug Resistance Network (CHAIN) grant number 223131.
Author information
Authors and Affiliations
Corresponding author
Additional information
E. Heger and A. Thielen contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
430_2011_199_MOESM2_ESM.doc
Supplementary Figure 1: Triplet and amino acid (aa) distribution in R5 and X4 strains. V3 sequences with defined phenotypes from the Los Alamos Database were analysed. According to the amino acid alignment, nucleotides were aligned and compared for their appearance in R5 (black) and X4 (grey). The percentage of each triplet (adenosine (abbreviated A), cytosine (C), guanine (G) and thymine (T) and the corresponding amino acid (Ala, alanine; Arg, arginine; Asn, aspargine; Asp, aspartic acid; Gln, glutamine; Glu, glutamic acid; Gly, glycine; Lys, lysine; Ser, serine; Thr, threonine, supplementary Table 1) for the position 11, 22, 24, and 25 are shown. Amino acids are grouped with respect to their physicochemical characteristics. (DOC 187 kb)
430_2011_199_MOESM3_ESM.doc
Supplementary Figure 2: G-to-A changes from R5 to X4 strains show highest correlation. The differences at each position in V3 between R5 and X4 strains regarding the percentage of nucleotide pairs (adenosine (abbreviated A), cytosine (C), guanine (G) and thymine (T) from the HIV-GRADE (A) and the Los Alamos (B) data set were calculated and correlated. Based on this calculation, the positions with the highest differences in R5 vs. X4 that mutate from one nucleotide to the other are located in the fourth (lower right) quadrant. The greatest correlation is boxed in red and enlarged (x ≥ 0.05, y ≤ -0.03). Nucleotide and triplet positions are indicated and separated by a slash. (DOC 1289 kb)
Rights and permissions
About this article
Cite this article
Heger, E., Thielen, A., Gilles, R. et al. APOBEC3G/F as one possible driving force for co-receptor switch of the human immunodeficiency virus-1. Med Microbiol Immunol 201, 7–16 (2012). https://doi.org/10.1007/s00430-011-0199-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-011-0199-9