
Abstract
Early detection of bloodstream infections (BSI) is crucial in the clinical setting. Blood culture remains the gold standard for diagnosing BSI. Molecular diagnostic tools can contribute to a more rapid diagnosis in septic patients. Here, a multiplex real-time PCR-based assay for rapid detection of 25 clinically important pathogens directly from whole blood in <6 h is presented. Minimal analytical sensitivity was determined by hit rate analysis from 20 independent experiments. At a concentration of 3 CFU/ml a hit rate of 50% was obtained for E. aerogenes and 100% for S. marcescens, E. coli, P. mirabilis, P. aeruginosa, and A. fumigatus. The hit rate for C. glabrata was 75% at 30 CFU/ml. Comparing PCR identification results with conventional microbiology for 1,548 clinical isolates yielded an overall specificity of 98.8%. The analytical specificity in 102 healthy blood donors was 100%. Although further evaluation is warranted, our assay holds promise for more rapid pathogen identification in clinical sepsis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Blood culture, which currently remains the gold standard in the microbiological diagnosis of bacterial or fungal bloodstream infections (BSI), typically becomes positive 8–36 h after sampling, and therapy can then be adapted based on presumptive bacterial identification suggested by Gram-stain characteristics. A more precise pathogen identification and susceptibility profile, however, is not available until up to 24–48 h [1, 2]. For the majority of patients with clinically apparent sepsis, the blood culture is negative, making the optimal antimicrobial therapy empiric [3]. Early detection and adequate treatment of causative pathogens within the first 6–12 h, however, is critical for a favorable outcome in patients with BSI [4–6]. The development of rapid diagnostic methods for BSI has been identified as an important medical need to supplement conventional blood culture diagnostics and molecular techniques have potential to fulfill this need [1]. Nucleic acid based diagnostic systems, including polymerase chain reaction (PCR) methods as well as the application of DNA and RNA probes are well known sensitive techniques for a more rapid detection and the specific identification of pathogens involved in BSI [7–15]. From a theoretical point of view, PCR-based diagnostic techniques, therefore, hold promise for sensitive and specific detection of target pathogens within much shorter turn around times than usually exercised with conventional blood cultures.
Microbiological and epidemiological data clearly indicate that only a limited number of bacteria and fungi are responsible for the majority of all bloodstream infections on the intensive care unit (ICU). As revealed in recent studies, about 20–25 species account for >90% of all detected nosocomial pathogens in BSIs [16, 17]. To correctly identify a pathogen in a patient with clinical sepsis and to cover the broad spectrum of microorganisms involved in BSI, several parallel or serial specific PCR analyses or a more universal PCR assay followed by specific probe hybridization or sequencing of the genomic target must be performed. Such an approach, however, is technically challenging, laborious, and time-consuming. Consequently, only PCR-based methods designed to specifically detect single pathogens (i.e., PCR for certain viruses) have been widely adopted in the routine clinical laboratory [18]. To date, several attempts have been made to translate the more universal diagnostic approach of blood-cultures into PCR-based assays that provide broad diagnostic coverage for the most frequent nosocomial pathogens involved in BSI [3, 15, 19, 20]. This has been done by the use of universal primers for PCR amplification that target conserved and specific genomic regions of pathogens and subsequent analysis of the PCR amplicon for pathogen identification by melting point analysis, hybridization probes, or direct sequencing [3, 20]. However, most of the broad range PCR assays introduced so far, still suffer from important technical limitations: preparation is done from plasma and not from whole blood, thus, missing bacterial cells or bacterial DNA already engulfed by phagocytic cells such as granulocytes and macrophages. Moreover, fungi are usually not part of such pathogen panels and most assays lack process controls to check for correct DNA preparation and PCR amplification. Finally, such assays commonly lack application of efficiently decontaminated reagents or alternatively, the use of high quality DNA-free reagents and plastic ware, to avoid potential workflow contamination resulting in impaired test sensitivity and specificity. To circumvent current technical limitations and to contribute to a more rapid detection and identification of pathogens in patients with clinical sepsis, here, we describe the development of a new sensitive and specific multiplex real-time PCR-based assay for amplification of bacterial and fungal DNA directly from whole blood samples.
Materials and methods
Assay workflow
The assay follows three steps: (1) specimen preparation by mechanical lysis and purification of DNA, (2) Real-time PCR amplification of target DNA in three parallel reactions (Gram positive, Gram negative, fungi) and subsequent detection of PCR products by specific hybridization probes, and (3) automated identification of species and controls. All specimen handling preparations and the PCR reaction set up were performed in a laminar flow box to minimize the risk of workflow contamination. Moreover sterile powder free gloves were worn. All instruments used were decontaminated from bacterial DNA prior to performance of the assay either by UV radiation or by decontamination reagents (e.g., LTK 008).
Specimen preparation
Samples of 3 ml whole blood specimens, were subjected to mechanical lysis with ceramic beads in a MagNALyzer® instrument (Roche Diagnostics GmbH, Mannheim, Germany) for 30 s at 6,500 rpm. After lysis, bacterial DNA was prepared using the SeptiFast Prep Kit Mgrade (Roche, Diagnostics GmbH, Mannheim, Germany) according to the manufacturer’s recommendations. Briefly, the lysed specimens were incubated at 56°C for 15 min with gentle agitation with a protease and chaotrophic lysis buffer (containing 50% guanidinium thiocyanate, 20% Triton X-100, 2% Proteinase K) that releases the nucleic acid and protects the released DNA from DNAses in the whole blood. Internal control (IC; 10 μl) was introduced into each specimen along with the lysis buffer. The IC consists of a mixture of synthetic double stranded DNA molecules with primer binding sites identical to those of the target sequences but differing in their hybprobe binding sites, thus allowing differentiation of the amplified IC and the target-specific amplicon. After addition of binding buffer the mixture was transferred to a spin column with a glass fiber insert (1,900g; 3 min). Human genomic DNA and the bacterial/fungal target DNA in the sample were then adsorbed to the surface of the glass fiber. Unbound substances (salts, proteins, cellular fragments) were removed by a washing step (4,200g; 2 min) with 1,800 μl of inhibitor removal buffer (containing 50% guanidinium thiocyanate, 40% ethanol, Tris–HCl buffer) and a subsequent washing step (4,200g, 10 min) with 1,600 μL of wash buffer (containing 0.2% sodium chloride, 80% ethanol, Tris–HCl buffer). Adsorbed nucleic acids were then eluted from the column after incubation (5 min) with 100 μL preheated (70°C) elution buffer and centrifuged for 2 min at 4,200g.
PCR amplification on the LightCycler® 2.0 Instrument
The eluate was subjected to three different PCR reactions using the LightCycler® SeptiFast Kit Mgrade (Roche Diagnostics GmbH, Mannheim, Germany) as recommended by the manufacturer. Briefly, three 50 μL aliquots of the prepared specimen DNA were added to the master mix and loaded onto the LightCycler capillaries (100 μl). The target DNA was amplified in three parallel reactions using the Hot start Taq-Polymerase. Gram positive, Gram negative or fungal sequences were amplified in the corresponding capillaries according to the bacterial and fungal master list (Table 1). The internal transcribed spacer region (ITS) was selected as specific target for the detection of bacterial and fungal pathogens. Sequence information of selected ITS target sequences is disclosed in patent applications WO 2004/052606, WO 2004/053156, WO 2005/075673, and WO 2006/035062. The target sequences are located between the 16S and the 23S ribosomal DNA sequences of Gram negative and Gram positive bacteria and between the 18S and 5.8S ribosomal sequences of fungi. The target sequences of the different species covered by the assay were amplified either with universal or specific primers. The assay principle including applicable primer and hybridization probe sequences of the ITS target regions is disclosed in patent applications WO 2004/053148 and WO 2004/053156. More detailed PCR-amplification conditions are outlined in Table 2.
Real time detection of PCR products by hybridization probes
During PCR reaction, increase of specific products was determined using dye-labeled hybridization probes and continuous automated fluorescence measurement. After completion of amplification, melting curve analysis was performed to further prove specificity of the products. The melting curve profile is given in Table 2. The probes hybridize to internal sequences of the amplified fragments during the annealing phase. Emitted fluorescence was then measured in one of four detection channels of the LightCycler® 2.0 Instrument. The melting temperature depended upon fragment length, composition of sequence, and degree of homology between the hybridization probe and the target DNA. All probes were designed to reliably discriminate between the different species as detected in the same detection channel of the instrument by use of specific melting temperatures of the corresponding amplicons. Examples of melting curve registrations and subsequent specific identification of microorganism, and internal controls by melting point analysis of the PCR-products are given in Fig. 1a–c. The corresponding specific melting temperatures for all PCR products and probes as derived from the respective pathogens are given in Fig. 2a–c.

Examples of characteristic melting curve registrations of spiking experiments and microorganism identification by respective melting temperatures. a K. oxytoca (T m = 61°C), b S. aureus (T m = 62°C), c C. albicans (T m = 55°C), and internal control (T m = 46°C)

Distribution of melting temperatures (T m) and respective detection channels for all microorganisms and internal controls. a Gram negative bacteria, b Gram positive bacteria, and c fungi
Automated identification of species and controls
Amplicons resulting from PCR reactions in specimens and controls were analyzed by a pathogen identification software (SIS; SeptiFast Identification Software, Roche Diagnostics GmbH, Mannheim, Germany) specifically designed for the interpretation of melting temperatures of PCR products. After the completion of each run on the LightCycler® 2.0 Instrument, all melting curves were marked manually with vertical sliders. Once a slider was set, the software automatically calculated the melting point (T m) value and the corresponding peak height. Specific T m boundaries had been pre-defined for each melting peak maximum. This procedure was repeated for each of the four detection channels and with all the three assays (Gram+, Gram-, Fungi; Fig. 2a–c). Minimal peak heights for all specified melting peaks were derived from a total of 13,727 data sets obtained with target negative specimen.
Preparation of blood specimen spiked with bacterial and fungal reference strains
Spiked specimen were prepared by spiking target-negative K-EDTA blood from healthy volunteers without clinical and laboratory evidence of infection with quantified cryostock preparations from bacterial or fungal reference strains to a final concentration of 100, 30, and 3 CFU/ml respectively. The list of reference strains used throughout the evaluation is given in Table 3. Cryostocks were prepared from conventional microbiological cultures of reference organisms. Aliquots of the prepared cryostocks were then stored at –80°C until use. Concentration of microorganisms were determined and adjusted by standard counting of colony forming units (CFU) after plating suspensions on solid media.
Evaluation of assay precision
The assay precision as measured in each detection channel was evaluated using pre-prepared K-EDTA whole blood (see above) spiked with 103 and 102 CFU/ml of microorganisms representative for each of the four detection channels of the LightCycler® 2.0 Instrument. S. marcescens was used for the 610 nm channel, K. oxytoca was used for the 640 nm channel, C. krusei was used for the 670 nm channel, and E. faecalis was used for the 705 nm channel. Specimens were analyzed in replicates of seven for intra-assay precision using six different instruments. For inter-assay precision, six independent experiments were performed. Lot-to-lot variability was investigated by including two different test lots.
Determination of minimal analytical sensitivity and specificity (inclusivity/exclusivity)
The assay minimal analytical sensitivity for each of the 25 species of the master list was analyzed by hit rate analysis. Target negative K-EDTA blood specimen of healthy donors (see above) serially spiked with bacterial and fungal reference strains (Table 3) to a final concentration of 100, 30, and 3 CFU/ml were then analyzed for the presence of the corresponding microorganisms in replicates of 8, 12, and 20, respectively. To guarantee assay inclusivity for different strains of the pathogens in the clinical setting, the assay was tested on a collection of 1,548 clinical isolates originating from various geographic areas in Europe (northern, central, and southern Europe) and US. All isolates were microbiologically characterized by phenotypical and biochemical methods and analyzed with the PCR test. In addition, 31 clinically common species not related to blood stream infections (Table 4) were tested for exclusivity of the assay by spiking of target negative K-EDTA blood samples and subsequent hit rate analysis.
Introduction of CP cut-offs and performance evaluation of analytical assay specificity
In first step, serial semi quantitative analysis of 574 blood samples obtained from a fresh veni-puncture site in 259 healthy volunteers according to common guidelines [21] was performed to determine the relative bacterial loads and positivity rates in such blood specimens of coagulase negative Staphylococci (ConS) and Streptococcus spp. upon application of semi-automated real-time PCR. These experiments aimed at establishing optional semi quantitative crossing point (cp) cut-off values for the LightCycler® 2.0 Instrument to potentially discriminate between significant bacterial loads of such organisms in clinical samples and low amounts of bacterial DNA (background reactivity) resulting from cross-contamination of samples by colonizing skin flora during the initial blood draw.
In second step, the previously established semi quantitative cp cut-off values were then evaluated by processing whole blood samples that had been collected from 102 individuals at two independent study sites under routine conditions of high throughput ICUs after approval of the local ethics committee and acquisition of written informed consent. To simulate blood sampling in ICU patients with indwelling devices undergoing invasive measures in the ICU, the blood draw was performed in individuals scheduled for elective cardiothoracic surgery. No clinical or laboratory signs of infection were obvious in these patients upon inclusion in the study. Blood cultures and EDTA specimens for PCR were collected in parallel directly from central lines or indwelling arterial catheters that had been inserted immediately before the blood draw using sterile precautions and after appropriate skin disinfection. Conventional blood cultures and PCR samples were then analyzed in parallel as described above at both study sites. Moreover, all PCR results were further re-analyzed for potential cross-contaminating skin flora by examining the raw data without using the automatic software threshold.
Statistics
Data was summarized and described by standard descriptive statistical methods (e.g., mean, standard deviation, variance, etc.) using Excel 2003 for Windows. Analysis of differences between melting peak temperatures of different isolates of the same species was done by Mann–Whitney U test using GraphPad Prizm 3.0.
Results
DNA extraction and assay performance
The DNA of all 25 bacterial and fungal species (Table 1) was successfully extracted directly from spiked whole blood samples without any preincubation and samples were then further analyzed by PCR using DNA free reagents. Microorganisms of each group (Gram positive, Gram negative, fungi) as well as the internal controls were identified by their specific melting points (T m) in the predefined detection channels of dedicated wavelength. Examples of melting curve registrations of typical spiking experiments and consecutive microorganism identification by respective melting temperature analysis are given for K. oxytoca (T m = 61°C; Fig. 1a), S. aureus (T m = 62°C; Fig. 1b) and C. albicans (T m = 55°C; Fig. 1c). Summarized information on detection wavelength and T m for each pathogen and the internal control is illustrated in Fig. 2a–c. Ambiguous T m were observed for several ConS species (51–58°C), but these could be clearly distinguished from the clinically important S. aureus (T m = 62°C; Fig. 2b). Moreover, E. aerogenes and E. cloacae yielded identical T m at 67°C (Fig. 2a) and were reported as E. aerogenes/cloacae.
Assay precision
Precision of the T m determination was tested for all detection channels of the LightCycler® 2.0 Instrument with four suitable reference organisms (S. marcescens, ATTC: 13880; K. oxytoca, ATTC: 13182; C. krusei, ATTC: 24210; E. faecalis, ATTC: 19433) at two different sample concentrations (1,000 and 100 CFU/ml) including two different test lots. Using seven replicates per sample on six different instruments a maximal intra-assay variance of 0.22°C (SD: 0.03–0.22°C) was observed. The maximum coefficient of variation calculated for T m was 0.38% (SD: 0.06–0.38%). More detailed results are given in Fig. 3a. The overall inter-assay precision calculated from six independent experiments varied between 0.19 and 0.54°C (SD: 0.12–0.27°C). The maximum coefficient of variation calculated for T m was 0.87% (SD: 0.37–0.87%). More detailed results are given in Fig. 3b.

Assay precision: precision of individual channels was tested with the following pathogens S. marcescens (channel 610), K. oxytoca (channel 640), C. krusei (channel 670), and E. faecalis (channel 705). The mean melting temperatures (T m) (±SD) for all results are presented. a Intra-assay precision was tested with 1,000 CFU open bars or 100 CFU grey bars of pathogen concentration for each detection channel in replicates of seven. b Inter-assay precision was tested with two different lots of bacteria open bars for lot A and grey bars for lot B, for each detection channel in six independent experiments. Data are given as mean ± SD
Determination of semi quantitative analytical cp cut-off values
Statistical analysis of experimental data as obtained from 574 blood samples that were drawn from a fresh veni-puncture site of 259 healthy volunteers after serial semi quantitative analysis by real time PCR revealed significant cross-contamination of the clinical samples in 32.7% for ConS and in 6.3% for Streptococcus spp. by colonizing skin flora. To reduce this positivity rate due to potential cross-contamination, application of a semi-quantitative analytical cp cut-off value was evaluated for ConS and Streptococcus spp. A cut-off at 25 cycles reduced the positivity rate to 22.4% for ConS and to 4.2% for Streptococcus spp.; a cut-off at 20 cycles resulted in a positivity rate of <0.5% for both pathogens. According to these experimental results a semi quantitative analytical cp cut-off value at 20 cycles for ConS and Streptococcus spp., seemed suitable to potentially discriminate between significant bacterial loads of such organisms in clinical samples and low amounts of bacterial DNA (background reactivity) resulting from cross-contamination by colonizing skin flora during the initial blood draw.
Determination of minimal analytical sensitivity and specificity
At concentrations of 100 CFU/ml the minimal analytical sensitivity of the assay, as revealed from analysis of EDTA-blood samples (n = 8/reference organism), was 100% for all reference strains tested (Table 3). In EDTA-blood samples (n = 12/reference organism) spiked with bacterial and fungal reference strains at a concentration of 30 CFU/ml hit rates were achieved between 70 and 100%, at concentration of 3 CFU/ml recorded hit rates were between 50 and 100% (Table 3). Due to the application of our predefined cross-contamination threshold the detection limit for ConS and Streptococcus spp. throughout our experiments was limited for several species and generally ranged between 50% for S. haemolyticus and 100% for S. pneumoniae (Table 3; Streptococcus spp. are denoted by an asterisk) at a concentration of 30 CFU/ml in the spiked samples. The hit rate rate for fungal agents varied substantially and ranged between 0% for C. glabrata and 100% for A. fumigatus in spiked samples at 3 CFU/ml (Table 3). Differentiation of microorganisms by the PCR test is based on different melting peak temperatures of the target-specific hybridization probes and the specific detection channel used. Evaluation of the melting peak temperatures of all 25 target microorganisms with bacterial and fungal reference strains revealed that the species differentiation specificity (ΔT m between target organisms detected in the same channel) was 3.37°C for P. mirabilis and S. marcesens and >5°C for all other target microorganisms, except for P. mirabilis and S. marcesens (ΔT m = 3, 37°C; Fig. 2a). No false positive samples were registered when assay specificity was tested. No significant differences in the melting peak temperatures were observed between different isolates of the same species (inclusivity; p > 0.05).
Uniformity and homogeneity of the ITS target region
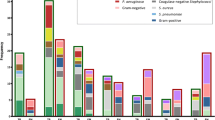
For each of the 25 microorganisms covered by the test, 13–158 different clinical isolates (1,548 isolates in total) were analyzed and compared to the result obtained by phenotypical and biochemical methods (Fig. 4). The overall sensitivity (number of detected isolates/number of isolates tested) is 98.8%, with an overall accuracy of 98.8% (number of correctly detected isolates/number of isolates detected). Discrepant results were found in 5 out of 128 Enterobacter strains which were found to have identical ITS regions to Klebsiella (pneumoniae/oxytoca), therefore, were reported as Klebsiella (pneumoniae/oxytoca). In addition, some Citrobacter freundii and Salmonella enterica isolates showed identical ITS regions to Klebsiella (pneumoniae/oxytoca) and E. coli, and therefore, were reported as Klebsiella (pneumoniae/oxytoca) and E. coli by the SIS, respectively. Moreover, due to sequence homologies 14 of 158 Streptococcus viridans strains were found identical to Streptococcus pneumoniae within the ITS region and were interpreted as S. pneumoniae. However, only 19 of 1,548 clinical isolates (1.2%) were found discrepant when PCR results and microbiological characterization of strains were compared. Finally, exclusivity was tested with ATCC strains of 31 clinically common microorganisms in triplicate in two different experiments (Table 4). No species tested for exclusivity was found positive by the assay (total of 186 negative screened samples).

Analytical inclusivity: 13–158 different clinical isolates were analyzed. Discrepant results were found in 5 out of 128 Enterobacter (cloacae/aerogenes) strains that were interpreted as Klebsiella (pneumoniae/oxytoca) due to sequence homologies. Streptococcus viridans (14 of 158) strains were interpreted as S. pneumoniae due to sequence homologies. In 14 samples no PCR amplification was possible. In total, 19 of 1,548 clinically isolates (1.2%) were found discrepant when comparing traditional phenotypic microbiological characterization and PCR identification
Performance test for diagnostic specificity in healthy volunteers
Whole blood samples of 102 patients scheduled for cardiothoracic surgery but without evidence of infection were analyzed by parallel examinations of conventional blood cultures and PCR samples at two independent study sites. ConS were detected in 20 patients by conventional blood cultures obtained directly from central lines or indwelling arterial catheters that had been inserted immediately before the blood draw using sterile precautions and after appropriate skin disinfection. All of the positive blood cultures (19.6%) yielded positive results 4–6 days after the blood draw, clearly pointing to cross contamination of the cultures with small amounts of residual skin flora. Interestingly, no pathogens were detected using the PCR system combined with subsequent SIS software analysis and activated threshold interpretation for ConS contamination again pointing to low levels of contaminating ConS in the corresponding blood culture positive samples. Re-evaluation of PCR datasets after disabling the established optional semi quantitative cp cut-off value for the LightCycler® 2.0 Instrument, however, showed that 33 (32.3%) ConS positive results could be generated from our sample set. In all these cases, the observed cp related to very low concentrations of pathogens as did the prolonged cultivation time in the positive blood cultures.
Discussion
The ultimate goal of using molecular technology for diagnosis of infection in the clinical setting is for the early detection of pathogens. Here, we introduce a novel molecular biological assay for parallel detection and identification of 25 clinically important BSI microorganisms. Most importantly, the total time-to-result including DNA preparation, PCR reaction, and data analysis is <6 h starting from a 3 ml K-EDTA whole blood specimen. The novel PCR assay does not require time-consuming pre-incubation of blood cultures prior to running the assay to increase analytical sensitivity. Compared to other PCR based diagnosis tools described previously, e.g., [20, 22] this system, includes external reaction controls to monitor proper function of test ingredients like primers and hybridization probes. Additionally, an internal process control is added to each individual clinical sample prior to DNA preparation. After amplification, the presence of specific amplicons is verified by use of specific dye-marked probes and semi-automated melting point analysis followed by software-assisted analysis to ensure high quality semi-automated species identification.
It has been previously reported, that in non-decontaminated PCR assays using universal primers, background levels of up to 1,000 copies of contaminating E. coli DNA can be present, thereby impairing the analytical sensitivity and specificity of such assays [23]. Such non-specific signals may arise from contaminations environmental microorganisms or by bacterial contamination of PCR ingredients. To avoid non-specific signals, and to increase analytical sensitivity, high quality PCR reagents, free of bacterial or fungal DNA contamination, were used according to suggestions recently discussed in the scientific literature [24, 25]. This represents a major improvement as application of conventional non-decontaminated PCR reagents usually show a background threshold of between 500 and 1,000 copies of bacterial target DNA/reaction [23, 25]. To increase analytical sensitivity even further, the internal transcribed spacer (ITS) region was selected as the target region for bacterial (16S–23S) and fungal (18S–5.8S) species identification. The ITS region offers higher analytical sensitivity compared to single copy targets since it is present in multiple operons in the genomes of bacteria and fungi [26]. Moreover, the ITS region is more species specific compared to conserved rDNA genomic region and therefore is more suitable for species differentiation [27].
In serial experiments performed on EDTA-blood samples spiked with different concentrations of bacterial and fungal reference organisms, hit rates of 70–100% were achieved for 23 out of 25 organisms at 30 CFU/ml and for 15 out of 25 organisms at 3 CFU/ml. The detection limit for ConS and Streptococcus spp. throughout our experiments was limited for several species due to the application of our predefined cross-contamination threshold and generally ranged between 50% for S. haemolyticus and 100% for S. pneumoniae. Similarly, the recovery rate for fungal pathogens varied substantially at concentrations of 3 CFU/ml and ranged between 0% for C. glabrata and 100% for A. fumigatus in the spiked samples (Table 3). The reason for the lower analytical sensitivity in C. glabrata positive samples clearly requires further investigations but may be caused by a reduced efficiency of the amplification reaction owing to the larger genomic target flanked by the primers designed for the ITS region in this specific organism.
The results of quantitative blood culture studies have shown that most episodes of clinically significant bacteraemia in adults are characterized by low numbers of circulating bacteria. In children, however, the magnitude of bacteraemia is usually much higher than in adults and in general it is inversely related to the child’s age. Werner et al. [28] found that 54% of blood cultures from adult patients with staphylococcal and streptococcal endocarditis contained between 1 and 30 CFU/ml of blood. Moreover, Kreger et al. [29] described that 73% of 77 patients with Gram negative bacteremia had blood cultures that contained <10 CFU/ ml of blood. An analytical assay sensitivity of 3–30 CFU/ml, however, clearly limits the diagnostic capabilities of our assay in patients with paucibacillary bacteremia when compared to a theoretical sensitivity of 1 CFU/culture bottle in conventional blood culture after inoculating ∼10 ml of whole blood. On the other hand, the clinical value of classical blood culture techniques is clearly impaired by the prolonged time-to-result interval, as it usually takes 1–3 days to provide the clinician with Gram-stain results and species identification. At present, our novel PCR based assay system, therefore, cannot fully substitute conventional blood culture due to its potentially lower sensitivity. However, it may provide valuable additional information to the treating physicians very early in the course of disease thereby potentially improving antibiotic coverage in septic patients.
Our system, in principle, permits rapid simultaneous detection of 25 different microorganisms using wide signal-to-signal melting peak differences (ΔT m = 3.36–5.0°C). Such specific and simultaneous identification of multiple target organisms requires the use of various specific primer and probe systems combined with multiple detection capabilities. In addition, the detection and differentiation of multiple organisms in a blood sample is strongly dependent on the amount of sequence diversity of the target regions in the individual microorganisms to then allow unambiguous differentiation by melting curve analysis after amplification. The overall assay precision for T m measurements of melting peak temperatures as determined for the representative microorganisms in all four detection channels of the LightCycler® 2.0 Instrument, varied between 0.19 and 0.54°C only. The differences in the melting peak temperatures was 3.37°C for P. mirabilis and S. marcescens and >5°C for all other 23 target organisms. Given the maximal temperature variance of 0.54°C, the observed signal-to-signal difference allowed unambiguous differentiation of all organisms of the master list as detected by the new real-time PCR-based assay system (Fig. 1a–c). In summary, this is a clear improvement compared to recently published PCR-based pathogen detection methods allowing detection and differentiation of up to 17 organisms only while using narrow signal-to-signal melting peak differences (ΔT m = 0.5–1.0°C) [20, 22].
In another evaluation step, the diagnostic specificity of our assay was assessed by testing the performance in the clinical setting at two independent study sites. To simulate blood sampling in individuals with indwelling devices on the ICU, blood cultures and EDTA-blood specimens for PCR were collected from 102 patients scheduled for cardiothoracic surgery. Samples were taken in parallel directly from central lines or indwelling arterial catheters that had been inserted to the patients immediately before the blood draw under sterile precautions. In 20 out of 102 individuals, blood cultures (19.6%) yielded a ConS positive result after 4–6 days as pointing to cross contamination of cultures with small amounts of residual skin flora. Correspondingly, re-evaluation of PCR data sets after disabling the established optional semi quantitative crossing point cut-off value for the LightCycler® 2.0 Instrument showed that 33 (32.3%) positive results for ConS could be generated from our sample set. In all these cases, the observed cps related to very low concentrations of pathogens, as did the prolonged cultivation time in the positive blood cultures. Interestingly, no ConS positive results, however, were detected by our PCR assay when our semi quantitative crossing point cut-off was applied for software-assisted evaluation of the PCR results, suggesting superior specificity of the assay compared to conventional blood culture under the conditions of our study.
Novel real-time PCR-based molecular biological assays for rapid detection of important pathogens causing BSI can potentially overcome some of the limitations of conventional culture based microbiological techniques. As revealed by many clinical and microbiological studies [4, 30, 31] patient outcome is greatly improved with the early administration of antibiotic therapy. Moreover, microbiological and epidemiological data clearly indicate that a limited number of bacteria and fungi are responsible for the majority of all bloodstream infections on the ICU. About 25 species account for more than 90% of all detected nosocomial pathogens in BSIs [16, 17]. For our assay the number of detectable target microorganisms was thus limited to the 25 most relevant microorganisms thereby covering around 90% of the most common pathogens causing BSI. Our assay at present does not include molecular detection of resistance genes and therefore, cannot substitute conventional blood culture with subsequent susceptibility testing. Detection and species differentiation of microorganisms from the blood of sepsis patients within 6 h after the blood draw however, can provide the treating physician with important complementary information to better tailor initial empiric broad spectrum antimicrobial therapy and to avoid potential gaps in antibiotic coverage of such patients. Moreover, in a second diagnostic step additional PCRs can be performed to detect resistance genes such as vanA, vanB, or mecA, if infection with more resistant pathogens is suspected from the result of the initial PCR [3, 20].
In conclusion, our newly developed real-time PCR assay may be a valuable tool for more rapid diagnosis of pathogens involved in BSI from whole blood without requiring pre-incubation of cultures. Analytical performance data obtained throughout our methodological study indicate good to excellent laboratory detection and differentiation capabilities for 25 bacterial and fungal microorganisms that are known to be most relevant in patients with BSI´s. Furthermore, the assay demonstrated excellent analytical specificity under the conditions of a high throughput ICU in patients with indwelling devices. At this point, our assay is designed to supplement conventional blood cultures due to its higher diagnostic rapidity. Whether the assay will indeed provide higher clinical sensitivity and specificity as well as more reliable identification of microorganisms early on in the course of disease compared to classical blood culture technique and whether it may significantly contribute to a more rapid initiation of better-tailored antimicrobial therapy and improved cost effective patient management in conjunction with other laboratory markers, however, awaits further evaluation of the test in clinical studies on sepsis patients.
References
Struelens MJ, de Mendonca R (2001) The emerging power of molecular diagnostics: towards improved management of life-threatening infection. Intensive Care Med 27(11):1696–1698
Harbarth S, Garbino J, Pugin J, Romand JA, Lew D, Pittet D (2003) Inappropriate initial antimicrobial therapy and its effect on survival in a clinical trial of immunomodulating therapy for severe sepsis. Am J Med 115(7):529–535
Wellinghausen N, Wirths B, Franz AR, Karolyi L, Marre R, Reischl U (2004) Algorithm for the identification of bacterial pathogens in positive blood cultures by real-time LightCycler polymerase chain reaction (PCR) with sequence-specific probes. Diagn Microbiol Infect Dis 48(4):229–241
Ibrahim EH, Sherman G, Ward S, Fraser VJ, Kollef MH (2000) The influence of inadequate antimicrobial treatment of bloodstream infections on patient outcomes in the ICU setting. Chest 118(1):146–155
Morrell M, Fraser VJ, Kollef MH (2005) Delaying the empiric treatment of candida bloodstream infection until positive blood culture results are obtained: a potential risk factor for hospital mortality. Antimicrob Agents Chemother 49(9):3640–3645
Garnacho-Montero J, Garcia-Garmendia JL, Barrero-Almodovar A, Jimenez-Jimenez FJ, Perez-Paredes C, Ortiz-Leyba C (2003) Impact of adequate empirical antibiotic therapy on the outcome of patients admitted to the intensive care unit with sepsis. Crit Care Med 31(12):2742–2751
Davis TE, Fuller DD (1991) Direct identification of bacterial isolates in blood cultures by using a DNA probe. J Clin Microbiol 29(10):2193–2196
Carroll KC, Leonard RB, Newcomb-Gayman PL, Hillyard DR (1996) Rapid detection of the staphylococcal mecA gene from BACTEC blood culture bottles by the polymerase chain reaction. Am J Clin Pathol 106(5):600–605
Jansen GJ, Mooibroek M, Idema J, Harmsen HJ, Welling GW, Degener JE (2000) Rapid identification of bacteria in blood cultures by using fluorescently labeled oligonucleotide probes. J Clin Microbiol 38(2):814–817
Jordan JA, Durso MB (2000) Comparison of 16S rRNA gene PCR and BACTEC 9240 for detection of neonatal bacteremia. J Clin Microbiol 38(7):2574–2578
Kempf VA, Trebesius K, Autenrieth IB (2000) Fluorescent in situ hybridization allows rapid identification of microorganisms in blood cultures. J Clin Microbiol 38(2):830–838
Laforgia N, Coppola B, Carbone R, Grassi A, Mautone A, Iolascon A (1997) Rapid detection of neonatal sepsis using polymerase chain reaction. Acta Paediatr 86(10):1097–1099
Newcombe J, Cartwright K, Palmer WH, McFadden J (1996) PCR of peripheral blood for diagnosis of meningococcal disease. J Clin Microbiol 34(7):1637–1640
Shang S, Chen Z, Yu X (2001) Detection of bacterial DNA by PCR and reverse hybridization in the 16S rRNA gene with particular reference to neonatal septicemia. Acta Paediatr 90(2):179–183
Yang S, Lin S, Kelen GD, Quinn TC, Dick JD, Gaydos CA, Rothman RE (2002) Quantitative multiprobe PCR assay for simultaneous detection and identification to species level of bacterial pathogens. J Clin Microbiol 40(9):3449–3454
Biedenbach DJ, Moet GJ, Jones RN (2004) Occurrence and antimicrobial resistance pattern comparisons among bloodstream infection isolates from the SENTRY antimicrobial surveillance program (1997–2002). Diagn Microbiol Infect Dis 50(1):59–69
Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB (2004) Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39(3):309–317
Louie M, Louie L, Simor AE (2000) The role of DNA amplification technology in the diagnosis of infectious diseases. CMAJ 163(3):301–309
Sleigh J, Cursons R, La Pine M (2001) Detection of bacteraemia in critically ill patients using 16S rDNA polymerase chain reaction and DNA sequencing. Intensive Care Med 27(8):1269–1273
Klaschik S, Lehmann LE, Raadts A, Book M, Gebel J, Hoeft A, Stuber F (2004) Detection and differentiation of in vitro-spiked bacteria by real-time PCR and melting-curve analysis. J Clin Microbiol 42(2):512–517
Seifert H (2001) MiQ: qualitätsstandards in der mikrobiologisch-infektiologischen diagnostik; Heft 3: Sepsis-Blutkulturdiagnostik. Elsevier, Urban and Fischer
Wellinghausen N, Wirths B, Essig A, Wassill L (2004) Evaluation of the hyplex bloodscreen multiplex PCR-enzyme-linked immunosorbent assay system for direct identification of Gram-positive cocci and Gram-negative bacilli from positive blood cultures. J Clin Microbiol 42(7):3147–3152
Meier A, Persing DH, Finken M, Bottger EC (1993) Elimination of contaminating DNA within polymerase chain reaction reagents: implications for a general approach to detection of uncultured pathogens. J Clin Microbiol 31(3):646–652
Corless CE, Guiver M, Borrow R, Edwards-Jones V, Kaczmarski EB, Fox AJ (2000) Contamination and sensitivity issues with a real-time universal 16S rRNA PCR. J Clin Microbiol 38(5):1747–1752
Klaschik S, Lehmann LE, Raadts A, Hoeft A, Stuber F (2002) Comparison of different decontamination methods for reagents to detect low concentrations of bacterial 16S DNA by real-time-PCR. Mol Biotechnol 22(3):231–242
Gurtler V, Stanisich VA (1996) New approaches to typing and identification of bacteria using the 16S-23S rDNA spacer region. Microbiology 142(Pt 1):3–16
Barry T, Glennon CM, Dunican LK, Gannon F (1991) The 16s/23s ribosomal spacer region as a target for DNA probes to identify eubacteria. PCR Methods Appl 1(2):149
Werner AS, Cobbs CG, Kaye D, Hook EW (1967) Studies on the bacteremia of bacterial endocarditis. JAMA 202(3):199–203
Kreger BE, Craven DE, Carling PC, McCabe WR (1980) Gram-negative bacteremia. III. Reassessment of etiology, epidemiology and ecology in 612 patients. Am J Med 68(3):332–343
Weinstein MP, Towns ML, Quartey SM, Mirrett S, Reimer LG, Parmigiani G, Reller LB (1997) The clinical significance of positive blood cultures in the 1990s: a prospective comprehensive evaluation of the microbiology, epidemiology, and outcome of bacteremia and fungemia in adults. Clin Infect Dis 24(4):584–602
Kumar A, Roberts D, Wood KE, Light B, Parrillo JE, Sharma S, Suppes R, Feinstein D, Zanotti S, Taiberg L, Gurka D, Kumar A, Cheang M (2006) Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 34(6):1589–1596
Author information
Authors and Affiliations
Corresponding author
Additional information
Lutz Eric Lehmann and Klaus-Peter Hunfeld contributed equally to this work.
Rights and permissions
About this article
Cite this article
Lehmann, L., Hunfeld, KP., Emrich, T. et al. A multiplex real-time PCR assay for rapid detection and differentiation of 25 bacterial and fungal pathogens from whole blood samples. Med Microbiol Immunol 197, 313–324 (2008). https://doi.org/10.1007/s00430-007-0063-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-007-0063-0