
Abstract
Effects of withdrawal from ethanol drinking in chronic intermittent ethanol vapor (CIE)-exposed dependent rats and air-exposed nondependent rats on proliferation and survival of progenitor cells in the hippocampus and the medial prefrontal cortex (mPFC) were investigated. Rats were injected with 5′-Bromo 2-deoxyuridine 72 h post-CIE to measure proliferation (2 h-old cells) and survival (29-day-old cells) of progenitors born during a time-point previously reported to elicit a proliferative burst in the hippocampus. Hippocampal and mPFC brain-derived neurotrophic factor (BDNF) and tropomyosin-related kinase B receptor (TrkB) expression were measured 3 h or 21d post-CIE to evaluate neurotrophic signaling during a time point preceding the proliferative burst and survival of newly born progenitors. CIE rats demonstrated elevated drinking compared to nondependent rats and CIE rats maintained elevated drinking following protracted abstinence. Withdrawal from CIE increased BDNF levels in the hippocampus and mPFC, and subsequently increased proliferation in the hippocampus and mPFC compared to nondependent rats and controls. Protracted abstinence from CIE reduced BDNF expression to control levels, and subsequently reduced neurogenesis compared to controls and nondependent rats in the hippocampus. In the mPFC, protracted abstinence reduced BDNF expression to control levels, whereas increased oligodendrogenesis in dependent rats compared to nondependent rats and controls. These results suggest a novel relationship between BDNF and progenitors in the hippocampus and mPFC, in which increased ethanol drinking may alter hippocampal and cortical function in alcohol dependent subjects by altering the cellular composition of newly born progenitors in the hippocampus and mPFC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alcohol use disorder affects about 17 million adults in the United States (SAMHSA 2013), and is associated with neurological deficits including, but not limited to, loss of brain volume and cognitive impairments, particularly, in functions dependent on the hippocampus and frontal cortex (Sullivan et al. 2000a, b; Bechara et al. 2001; Oscar-Berman and Marinkovic 2003; Rosenbloom et al. 2003). Several widely accepted rodent models of alcohol dependence also implicate structural and functional impairments of the hippocampus and prefrontal cortex (PFC) as neurological factors that contribute to alcoholism (Stephens and Duka 2008; Crews and Boettiger 2009; Crews and Nixon 2009; Koob and Volkow 2010). One such model, namely the chronic intermittent ethanol vapor exposure (CIE) model, implements daily cycles of intoxication via ethanol vapors and withdrawal to induce clinical signs of alcohol dependence, such as somatic withdrawal symptoms and escalated ethanol drinking in rats (Valdez et al. 2002; O’Dell et al. 2004). Impairments in the hippocampus and medial prefrontal cortex (mPFC) function produced by CIE were associated with a suppression of neuroplastic mechanisms (e.g., neuronal activation, functional and structural neuronal plasticity, neurogenesis in the hippocampus and gliogenesis in the mPFC) during CIE in these regions (Richardson et al. 2009; Hansson et al. 2010; Pian et al. 2010; Badanich et al. 2011; Criado et al. 2011; George et al. 2012; Holmes et al. 2012; Kroener et al. 2012; Vendruscolo et al. 2012; Kim et al. 2015; Staples et al. 2015). However, few studies have investigated the alterations in neurogenesis in the hippocampus and gliogenesis in the mPFC and neurotrophic mechanisms in the hippocampus and mPFC during withdrawal and protracted abstinence from CIE (Hansson et al. 2010; Navarro and Mandyam 2015; Staples et al. 2015). Such studies may assist with understanding the neurobiological mechanisms underlying relapse to alcohol seeking behaviors evident in CIE animals during protracted abstinence (Liu and Weiss 2002; Vendruscolo et al. 2012).
In the context of the above hypothesis, human laboratory studies reveal that alcohol abstinence rescued, at least partially, the loss in brain volume in several brain regions (Demirakca et al. 2011; Durazzo et al. 2011; van Eijk et al. 2013), and that this recovery of brain volume was limited, or even absent, in individuals who had relapsed (Demirakca et al. 2011; Durazzo et al. 2011); preclinical studies may be useful for identifying associated mechanisms including structural and neuroplastic changes in neuronal and glial cells. For example, rodent studies demonstrate that the suppression of hippocampal cell proliferation following ethanol exposure is transient, and withdrawal from ethanol experience produces a rebound effect observed as increases in cell proliferation (Nixon and Crews 2004; Hansson et al. 2010). Few studies have investigated the capacity of the proliferating cells born during the rebound effect to survive and express neuronal and glial markers, therefore, the stability of the newly born cells during excessive proliferation is questionable. The current study tests the hypothesis that the development of hippocampal progenitors during withdrawal and protracted abstinence may be hindered due to a hostile microenvironment in the neurogenic niche. Additionally, the current study also tests the sub-hypothesis that proliferation and survival of mPFC progenitors (gliogenesis) is affected by withdrawal and protracted abstinence and these changes are associated with altered gliogenic niche.
The neurogenic niche in the hippocampus and the gliogenic niche in the mPFC are supported by the activity of trophic factors (Nestler 2002; Ming and Song 2005; Rajkowska and Miguel-Hidalgo 2007; Hagg 2009; Minichiello 2009; Gray et al. 2013). For example, brain-derived neurotrophic factor (BDNF) and its receptor tropomyosin-related kinase B (TrkB) are involved in the survival and differentiation of neural progenitor cells in the hippocampal granule cell layer (Scharfman et al. 2005; Bergami et al. 2008; Donovan et al. 2008). Similarly, BDNF and TrkB may assist with survival and differentiation of glial progenitor cells in the mPFC, particularly because TrkB receptors are expressed on oligodendroglial cells and modulate myelination (Wong et al. 2013). With respect to alcohol, converging evidence from various models of alcohol intoxication (CIE, intragastric intubation, two-bottle choice; liquid ethanol diet) demonstrate that ethanol exposure alters BDNF expression in the hippocampus and cortex (MacLennan et al. 1995; Tapia-Arancibia et al. 2001; Miller 2004; Miller and Mooney 2004; Logrip et al. 2009; Alele and Devaud, 2013; Briones and Woods 2013). Thus, dysregulated BDNF-TrkB signaling in the hippocampus and PFC may contribute to the maladaptive neuronal plasticity observed in alcohol dependent subjects in these brain regions (McGough et al. 2004; Russo et al. 2009). Therefore, the current study tested the hypothesis that abnormal BDNF-TrkB signaling contributes to the hostile environment promoting the rebound effect on proliferating cells in the hippocampus and mPFC.
Experimental procedures
Animals
Eighty-six adult male Wistar rats (Charles River, Hollister, CA USA) completed the study. The rats were housed two/cage in a temperature-controlled (22 °C) vivarium on a 12 h/12 h light/dark cycle (lights on 8:00 p.m. to 8:00 a.m.). All procedures were performed during the dark phase of the light/dark cycle. Food and water access was available ad libitum. All rats weighed approximately 220–250 g and were 8 weeks old at the beginning of the study. All experimental procedures were carried out in strict adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication number 85–23, revised 1996) and approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute.
Ethanol self-administration
Thirty-three experimentally-naïve rats were given two 14-h lever-responding training sessions in the operant conditioning boxes (Med Associates Inc, VT). During these sessions, rats were trained on fixed-ratio 1 schedule (FR1), where rats had access to one lever, and responding on the lever delivered 0.1 ml water into a single cup liquid receptacle placed between levers. Following the overnight water sessions, rats were trained for 4 days on 2-h FR1 schedule, where responding on the active lever delivered 0.1 ml ethanol (10 % v/v) and was followed by a 4-s time-out during which further responding on the active lever was recorded but did not result in the delivery of ethanol, and responding on the inactive lever was recorded but had no programmed consequences. Subsequently, the rats were trained on 30-min ethanol sessions on an FR1 schedule and maintained for the rest of the study.
During maintenance, after the rats reached stable responding (stable responding was defined as less than 10 % variation in active lever responding for 3 consecutive 30-min FR1 sessions), they were divided into two groups such that lever responses did not differ significantly between groups. One group received CIE (see procedure below; n = 15); henceforth, these rats are referred to as ‘dependent rats’. The second group (n = 18) were exposed to air in their normal housing condition (did not experience ethanol vapors), hence these rats are termed ‘nondependent rats’. During the last 5 weeks of the 7 week CIE exposure, the dependent and nondependent rats completed ethanol self-administration sessions twice a week (Tuesdays and Thursdays). Responding was analyzed to determine escalation of self-administration compared to pre-vapor stable responding. After 7 weeks of CIE, dependent rats were withdrawn from ethanol vapors and all rats were withdrawn from ethanol self-administration for 4 weeks. On the 28th and 29th day of abstinence, nondependent and dependent rats underwent 30-min ethanol self-administration session and were tested for relapse to drinking. The experimental design is presented as a detailed schematic in Fig. 1.

Schematic of experimental design demonstrating the animal groups used and the time points for behavioral and histological analysis. Adult male Wistar rats were trained to self-administer ethanol (10 % v/v) for 2 weeks in a 30-min operant conditioning paradigm. The rats were then divided into two groups; one group was exposed to the chronic intermittent ethanol vapor (CIE) paradigm and are referred to as dependent rats (n = 15 rats). The second group was not exposed to CIE, and are termed nondependent rats (n = 18 rats). Both groups were maintained on ethanol self-administration sessions twice a week; this continued for 7 weeks. At the end of the 7th week, somatic withdrawal behaviors were scored. Three days after the cessation of CIE exposure, rats were injected with BrdU (150 mg/kg; i.p.) to label newly born cells. Two hours after BrdU injections, nine dependent and nine nondependent rats were euthanized, and their brains were processed for immunohistochemical analysis of BrdU cells. The remaining rats were maintained in their home cages for 4 weeks with no exposure to ethanol vapor or ethanol self-administration (abstinence). On days 28 and 29 during abstinence, these rats underwent 30-min ethanol self-administration session. Immediately after the last session, rats were euthanized and their brains were processed for BrdU immunohistochemistry
Chronic intermittent ethanol vapor exposure (CIE)
During CIE, rat cages were housed in specialized chambers and exposed to a 14-h on/10-h off schedule for the ethanol vapors. Using a peristaltic pump (model QG-6, FMI Laboratory, Fluid Metering), 95 % ethanol from a large reservoir was delivered to a heated flask at a regulated flow rate. The drops of ethanol in the flask were immediately vaporized and carried to the vapor chambers containing the rat cages by controlled air flow (regulated by a pressure gauge). The air and ethanol flow rates were optimized to result in blood alcohol levels (BALs) between 125 and 250 mg/dl of or 27.2 and 54.4 mM (Gilpin et al. 2008a); these BALs are 2–3 times the BAL observed in binge drinking, but not high enough to abolish righting reflex (Ernst et al. 1976; Courtney and Polich 2009).
Tail bleeding for BAL measures
For measuring BALs, tail bleeding was performed on the dependent rats, once a week on the same day every week, during hours 13–14 of CIE (total 14 h exposure/day) according to (Gilpin et al. 2008b). Rats were gently restrained while the tip of the tail (2 mm) was cut off with a clean razor blade. Tail blood (0.2 ml) was collected and centrifuged. Plasma (5 μL) was used for measurement of blood alcohol levels (BALs) using an Analox AM1 analyzer (Analox Instruments USA Inc., MA, USA). Single-point calibrations were performed for each set of samples with reagents provided by Analox Instruments (25–400 mg/dl or 5.4–87.0 mM). When blood samples were outside the target range (125–250 mg/dl), vapor levels were adjusted accordingly. Mean BALs during the 7 week CIE exposure is reported in Table 1.
Scoring of physical withdrawal
At the end of vapor exposure during the 7th week, physical withdrawal was evaluated in dependent as well as nondependent rats. Nondependent rats did not reveal any physical withdrawal symptoms, hence were scored 0 on a scale of 0–3 for all behaviors. Data from nondependent rats is not presented. Data from dependent rats evaluated at two time points is presented. Two optimal time points were chosen for measuring physical withdrawal based on previous publications that have reported withdrawal behaviors hours to days after cessation of CIE (O’Dell et al. 2007; Gilpin et al. 2008a; Richardson et al. 2008). An early 2.5 h post-CIE time-point corresponded with the early onset of withdrawal, while the late 7 h post-CIE time-point corresponded with the peak of withdrawal following the termination of vapor exposure (O’Dell et al. 2007; Gilpin et al. 2008a; Richardson et al. 2008). Withdrawal signs in rats were scored by an observer blind to the dependence status of the rats using previously published rubric (Majchrowicz 1975; Macey et al. 1996). Individual withdrawal signs of abnormal body posture (“ridged posture of the entire body including limbs, associated with little flexibility and resilience to handling”; scored as: broad-based gait = 3, immobility = 2, and unsteady = 1 or slow = 0 forward locomotion), tail stiffness (“ridgedly extended in a horizontal plane” and “similar to an inflexible rod in contrast to a pliable and elastic tail found in untreated animals”; scored as: rigid tail that extends up toward the ceiling = 3; thirty to sixty degree angle = 2; parallel to the back of the rat = 1 or flexible tail = 0), hyperlocomotion (“frantic or frenzied exploration of the cage, excessive sniffing and locomotor activity, enhanced startle reflex, agitation and restlessness”; scored as: excessive running in circles = 3, moderate running in circles = 2, low running in circles = 1, spontaneous activity = 0), and stereotypic head movements (“repetitive shifting of the upper thorax and both forelimbs from side to ride or restless turning of the head from side to side” scored as: vigorous movement = 3, moderate movement = 2, low movement = 1, no repetitive movement = 0) were scored separately on a scale of 0–3 (Majchrowicz, 1975; Macey et al. 1996). Although the behaviors scored could reflect seizure-like activity, seizures were not measured and were not scored.
5′-Bromo 2-deoxyuridine (BrdU) Labeling
Three days following removal from ethanol vapor, dependent, nondependent, and control (both ethanol- and behaviorally-naïve; n = 12) rats received an injection of BrdU (150 mg/kg, i.p.). The 3 day time-point was selected based on a previous publication that reported a burst in proliferation in the hippocampal subgranular zone (SGZ) using a comparable CIE model (Hansson et al. 2010). The single BrdU dose (150 mg/kg) was chosen to maximize labeling of proliferating cells by using a near-saturating concentration of BrdU (saturating concentration in rodents is ~200 mg/kg). Of note, while BrdU is used for experimentally labeling proliferating cells (Dayer et al. 2003; Mandyam et al. 2007a; Taupin 2007), injections of BrdU are also indicated to have cytotoxic and teratologic effects (Kolb et al. 1999; Sekerkova et al. 2004; Ogawa et al. 2005; Kuwagata et al. 2007; Duque and Rakic 2011; Rowell and Ragsdale 2012), primarily because BrdU is a marker of DNA synthesis and not of cell division per se (Breunig et al. 2007). However, in adult rodent models, BrdU cytotoxicity is typically evident at greater than 2 times the currently used dose [(Cameron and McKay 2001; Eadie et al. 2005); for review, (Taupin, 2007)], suggesting that the BrdU dose used in the current study could be suitable to evaluate proliferation and survival of progenitors in adult rats.
To measure cellular proliferation of newly born cells, 2 h after the BrdU injection, a subset of dependent (n = 9), nondependent (n = 9), and control (n = 6) rats were euthanized, and their mPFC and hippocampal sections were processed for BrdU immunohistochemistry (Fig. 3b, e; detailed below). The remaining dependent rats (n = 6), nondependent rats (n = 9) and control rats (n = 6) were euthanized 29 days after BrdU injection to measure the survival of newly born cells in the hippocampus and mPFC (Fig. 4d, e). In the dentate gyrus, newborn cells that survive ~28 days are predominantly neuronal [~70 % become mature neurons, (Palmer et al. 2000)] and are stably incorporated into the granule cell layer (Kempermann et al. 2003). To that end, rats were maintained in their home cages for an additional 29 days without exposure to CIE or ethanol self-administration. On days 28–29, nondependent and dependent rats experienced 30-min self-administration session to evaluate relapse to drinking behavior. The rats were sacrificed 1–2 h after the drinking session and brain tissue was processed for BrdU immunohistochemistry to measure cell survival.
Immunohistochemistry and quantitative analysis of BrdU labelled cells
Rats were fully anesthetized using chloral hydrate (240 mg/kg, i.p.) and then transcardially perfused with phosphate-buffered saline (2 min at 15 ml/min) and 4 % paraformaldehyde (20 min at 15 ml/min). The brains were dissected out and postfixed in 4 % paraformaldehyde at 4 °C for 16–20 h, cryoprotected in 30 % sucrose, and cut into 40 µm sections along the coronal plane on a freezing microtome. Every ninth section through the PFC (anterio-posterior +3.7 to +2.5; 4 bilateral sections per rat and the hippocampus (anterio-posterior −2.1 to −4.2; 8 bilateral sections per rat) was mounted on Superfrost® Plus slides and dried overnight. Immunohistochemistry was performed using a previously published method (Mandyam et al. 2007b; Richardson et al. 2009) using rat monoclonal anti-BrdU (1:400; catalog # MCA2060, Serotec); the sections were counter-stained with Vector Fast Red (a nuclear stain).
BrdU labelled cells were quantified in the mPFC and the hippocampus with a Zeiss AxioImager Microscope equipped with Stereo Investigator 11.06 (MicroBrightField Bioscience, Williston, VT USA), a three-axis Mac 5000 motorized stage, a Zeiss digital MRc video camera, PCI color frame grabber, and computer work station. Hippocampal and mPFC regions were contoured using a 2.5× objective with a 10× eye piece and the above software. The mPFC was contoured by referencing histological landmarks including corpus callosum, anterior commissure and rhinal fissure while referring to the rat brain atlas and according to our previous publication (Paxinos and Watson 1997; Mandyam et al. 2007b). The hippocampal granule cell layer was contoured around the cellular portion of dentate gyrus (counterstained pink) that was morphologically distinct from the hippocampal hilus (colorless to pale-brown, for example refer micrographs in Figs. 3, 4). Cells were visually quantified and quantification of cells was restricted to within the contour using a 40× objective and a 10× eyepiece, following strictly outlined criteria (cells stained as dark brown to black, with the ability to focus the boundary of the cell within the mounted section thickness), and was performed by an observer blind to the study. A software generated 180 × 120 µm counting frame (optimized based on the complete visibility of the frame on the computer screen) was systematically moved through the entire contoured area of the tissue to manually assess and count the BrdU-positive (BrdU+) cells. Mounted section thickness after immunohistochemistry was determined to be ~28 µm and focus along the z-axis was adjusted continuously to define cell boundaries for visualizing BrdU+ cells. The 2-h-old BrdU+ cells in the SGZ [defined as a zone 50 um into the hilus with the edge starting from the hilar/granule cell layer border (granule cell layer (GCL) stained as packed nuclei and hilus, a layer without packed cells; Fig. 3a)] always appeared in clusters of irregularly shaped dark stained cells. The overlapping-pair arrangement and the number of cells in each cluster were confirmed by focusing on different layers of cells along the z-axis. Twenty-nine-day old BrdU+ cells in the granule cell layer were predominantly distributed as single cells or pairs of oval to round, spotty or dark stained cells, rarely seen in clusters, and exhibited cell morphology different from the proliferating time point (Fig. 4b). Two-hour-old BrdU+ cells in the mPFC also occurred in clusters or pairs of cells (Fig. 3d) and 29-day old BrdU+ cells in the mPFC were rarely seen in clusters (Fig. 4c). Absolute cell counting (complete counting of all immunoreactive cells in the contoured area) was performed in the mPFC and SGZ or granule cell layer and BrdU+ cells in the two hemispheres were summed for the both mPFC and hippocampus. BrdU+ cells in the hippocampus are presented as total number of cells per animal, and cells in the mPFC are presented as total number of cells per unit area (cells/mm2 based on mounted section thickness) per animal.
Phenotypic analysis of BrdU labelled cells
To determine the phenotype of 29-day-old BrdU+ cells, PFC and hippocampal sections were mounted on Superfrost slides as described above. Hippocampal sections were immunoprobed for BrdU (sheep primary: 1:500; Abcam, Cambridge, MA, horse anti-sheep secondary: 1:200), neuronal marker NeuN (mouse primary: 1:50; EMD Millipore, Billerica, MA, horse anti-mouse secondary: 1:200) and astroglial marker glial fibrillary acidic protein (GFAP; chicken primary: 1:500; Abcam, Cambridge, MA, donkey anti-chicken secondary: 1:200). PFC sections were immunoprobed for BrdU, GFAP and oligodendroglial marker Olig2 (rabbit primary: 1:100, goat anti-rabbit secondary: 1:200) (Kim et al. 2015). Confocal analysis of triple labeled cells in the granule cell layer and mPFC was performed with a Zeiss Axiovert 100 M and LSM510 using a previously published method (Kim and Mandyam 2014; Kim et al. 2015). Briefly, using a 60 × oil immersion objective (equipped with a 10 × eyepiece), immunoreactive cells were optically sectioned in the z-plane using multitrack scanning (section thickness of 0.45 μm). Colocalization of antibodies was assessed with the confocal system by analysis of adjacent z-sections (using gallery function and orthogonal function for equal penetration of the antibodies). The phenotypic distribution of the 29-day-old BrdU+ cells, was calculated as the percent of hippocampal BrdU+ cells colabeled with NeuN or GFAP, and as the percent of mPFC BrdU+ cells colabeled with Olig2 and GFAP in each rat. A minimum of 15 BrdU+ cells were analyzed per animal from each of the rats in the control (n = 6), dependent (n = 6) and non-dependent (n = 9) groups.
BDNF and TrkB Western blotting
A separate cohort of thirty-nine rats were used to determine whether changes in BDNF and TrkB protein expression were associated with altered cell proliferation and survival in the hippocampus and mPFC. Upon arrival to the animal facility, dependent rats (n = 21 rats) were exposed to the comparable CIE procedure as detailed above, where the rats were housed in ethanol vapor chambers for 7 weeks. The control rats (ethanol naïve, n = 18) were housed as described previously. To evaluate the effect of CIE exposure on BDNF signaling and the persistence of the CIE-induced changes during protracted abstinence, rats were decapitated under isoflurane anesthesia 3 h (acute withdrawal; n = 12) and 21 days (21d; protracted abstinence, n = 9), respectively, following cessation of ethanol vapor exposure. Brains were rapidly extracted and promptly snap frozen in dry ice-cooled isopentane and stored at −80 °C until further processing.
Western blot procedures optimized for measuring levels of both phosphoproteins and total proteins were employed (Graham et al. 2007; Kim et al. 2015; Galinato et al. 2015). Tissue punches from 500-um thick sections of mPFC and hippocampus were homogenized by sonication in ice-cold buffer (320 mM sucrose, 5 mM HEPES, 1 mM EGTA, 1 mM EDTA, 1 % SDS, with Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktails II and III diluted 1:100; Sigma, St. Louis, MO), and protein concentration was determined using a detergent-compatible Lowry method (Bio-Rad, Hercules, CA). Mature BDNF protein levels and TrkB and Tyr-706 phosphorylated TrkB (pTrkB) protein levels were determined from 30 µg protein samples subjected to gel electrophoresis, and transferred to PVDF membranes. Membranes were incubated with the appropriate primary antibody for 16-20 h at 4 °C: BDNF (1:200, SCBT sc-546), Tyr-706 pTrkB receptor (1:200, SCBT sc-135645), or total TrkB receptor (tTrkB, 1:200, SCBT sc-8316), and then with the appropriate horseradish peroxide–conjugated secondary antibody (goat anti-rabbit IgG, 1:200–500, or goat anti-mouse IgG1, 1:5000, BioRad) for 1 h at room temperature (24 °C). Immunoreactivity was detected using SuperSignal West Dura chemiluminescence detection reagent (Thermo Scientific) and collected using HyBlot CL Autoradiography film (Denville Scientific) and a Kodak film processor. Net intensity values were determined using the ImageStudio software (Li-Cor Biosciences). Blots were reprobed for β-Tubulin (1:8000, SCBT sc-53140) or stained with Coomassie Blue dye (Welinder and Ekblad, 2011) for normalization purposes. Protein expression is presented as percent of control samples on the same blot to normalize for blot-to-blot variability.
Statistical analysis
Ethanol self-administration during the 7-week CIE exposure was evaluated using repeated measures two-way ANOVA with weekly-session (or time) as a within-subject factor and CIE status as a between-subject factor. Significant interactions were investigated using Sidak’s post hoc tests. Ethanol self-administration during the 11th week was compared between the dependent and nondependent rats using unpaired t-tests. Both 2 h old and 29 day old BrdU+ cells in the mPFC and SGZ were compared between control, dependent and nondependent rats using one-way ANOVAs, followed by Tukey’s post hoc tests. Proliferation of BrdU cells in the mPFC and SGZ were correlated with ethanol self-administration levels using Pearson’s product-moment correlation coefficient. Triple-labeling analyses was conducted to compare hippocampal BrdU-only, BrdU and NeuN colabeled, and BrdU and GFAP colabeled cells between control, dependent and nondependent rats using two-way ANOVA. Similarly, PFC BrdU-only, BrdU and Olig2 colabeled, and BrdU and GFAP colabeled cells were compared between control, dependent and nondependent rats using two-way ANOVA. Main effects of cell-type in both hippocampus and PFC were probed using Tukey’s post hoc tests. Withdrawal scores at 2.5 h and 7 h post-CIE were compared using a non-parametric Wilcoxon matched-pairs signed rank test. Scores for the withdrawal signs were correlated with peak BALs and with BrdU-labeled cells in the mPFC and SGZ using Spearman’s rank correlation coefficient. For the Western blotting analyses, one-way ANOVAs followed by Tukey’s post hoc tests were used to compare values between acute withdrawal, protracted abstinence, and control groups. Statistical significance was accepted at p < 0.05.
Results
Escalation of ethanol self-administration
Responding for ethanol (10 % v/v) was not different between the groups of rats dedicated to the dependent and nondependent groups prior to the onset of vapor exposure [t (31) = 1.36; p = 0.09; Fig. 2a]. The amount of ethanol vapor experienced by each dependent rat reached the desired range by the second week of CIE exposure as indicated by the increases in BALs (Table 1). A significant time × CIE interaction was obtained (F[5,155] = 10.7, p < 0.001) for ethanol self-administration over the 7 weeks of CIE exposure. Further investigation of the interaction revealed that in the dependent rats, responding for ethanol increased from week 4–7 compared to pre-vapor responding (week-1; p’s < 0.01; Fig. 2a). No difference in responding was observed across weeks in the nondependent rats (p = 0.09). Also, from week 3 onwards, the dependent rats exhibited greater responding for ethanol compared to the nondependent rats (p’s < 0.001). During Week 7, BALs did not correlate with ethanol self-administration (r = −0.18, n.s.).

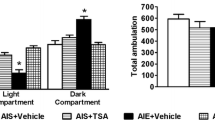
Escalation of ethanol self-administration during CIE and withdrawal scores following withdrawal from CIE. a Ethanol self-administration (active lever presses) increased over the 7 weeks of CIE exposure in the dependent rats but not in the nondependent rats. n = 15 and 18 for dependent and nondependent rats, respectively; *p < 0.05 compared to pre-vapor responding, within group; # p < 0.05 compared to non-dependent rats, between group. b Withdrawal behaviors increase from 2.5 to 7 h following removal of CIE exposure. Signs of ethanol withdrawal, i.e. aberrant body posture, tail stiffness and stereotypy as well as overall withdrawal signs increased in the dependent rats from early onset of withdrawal, at 2.5 h post-CIE, to peak onset withdrawal, at 7 h post-CIE. N = 15/group. *p < 0.05 compared to early withdrawal, within-subject
Responding for ethanol during week 11 remained significantly greater in the dependent rats compared to that observed in the nondependent rats (p < 0.05; Fig. 4a), suggesting that excessive responding for ethanol persisted following prolonged abstinence (i.e., after 4 weeks of abstinence).
Ethanol withdrawal
In dependent rats, withdrawal scores of body posture, tail stiffness, and stereotypic behavior increased between 2.5 h post-CIE to 7 h post-CIE (p’s < 0.05; Fig. 2b). Stereotypy in dependent rats at 7 h post-CIE correlated positively with peak BALs at week 7 (Table 2). Withdrawal scores did not correlate with ethanol self-administration or with cell proliferation in the SGZ or mPFC (Table 2).
Cell proliferation in the hippocampus and the mPFC
The number of 2 h-old BrdU+ cells was greater in the hippocampal SGZ of the dependent rats compared to both nondependent and control rats (F[2,21] = 4.11, p < 0.05; Fig. 3b). The number of BrdU+ cells in the SGZ of dependent rats positively correlated with ethanol self-administration during Week 7 of CIE (r = 0.74, p < 0.05, Fig. 3c). No such relationship was observed in the nondependent rats (r = −0.07, p = 0.9; Fig. 3c).

Withdrawal from CIE and ethanol drinking increases the number of proliferating cells in the SGZ and mPFC. a Photomicrograph of 2 h-old BrdU+ cells in the SGZ of the dentate gyrus. BrdU+ cells appeared in clusters; the cluster represented has 7 immunoreactive cells, each pointed with an arrowhead. In the hippocampus, the number of 2 h-old BrdU-labelled cells were greater in the dependent rats compared to both nondependent and control rats (b). The number of hippocampal BrdU cells positively correlated with ethanol self-administration during the 7th week of CIE, suggesting that greater ethanol drinking is predictive of reactive cell proliferation in the hippocampal SGZ (c). d Photomicrograph of 2 h-old BrdU+ cells in the mPFC. BrdU+ cells appeared in pairs; each immunoreactive cell is pointed with an arrowhead. In the mPFC, the number of 2 h-old BrdU-labelled cells were greater in the dependent rats compared to both nondependent and control rats (e). The number of mPFC BrdU cells did not correlate with ethanol self-administration during the 7th week of CIE (f). n = 6, 9, and 9 for control, nondependent, and dependent rats, respectively. *p < 0.05 compared to control rats; # p < 0.05 compared to nondependent rats. Scale bar in (d) is 10 µm, applies to (a, d)
In the mPFC, the number of 2 h-old BrdU+ cells were greater in the dependent rats compared to both nondependent and control rats (F[2,18] = 7.70, p < 0.01; Fig. 3e). However, cell proliferation in dependent and nondependent rat mPFC did not correlate with their ethanol self-administration during week 7 (r = 0.46, p = 0.2 and 0.68, p = 0.07, respectively Fig. 3f).
Cell survival in the hippocampus and the mPFC
In the hippocampal granule cell layer, the number of 29-day-old BrdU+ cells were fewer in the dependent rats compared to the nondependent rats (F[2,19] = 6.43, p < 0.01; Fig. 4d) but not compared to the control rats. In contrast, 29-day-old BrdU+ cells in the mPFC were greater in dependent rats compared to nondependent as well as the control rats (F[2,17] = 5.85, p < 0.05; Fig. 4e).

Cell survival is decreased in the hippocampus but increased in the medial prefrontal cortex of dependent rats during protracted abstinence. a Ethanol self-administration (active lever presses) following a protracted abstinence remained significantly greater in the dependent animals compared to nondependent rats. Photomicrographs of 29-day-old BrdU+ cells in the dentate gyrus granule cell layer (b) and mPFC (c). BrdU+ cells appeared as single cells or in pairs; each immunoreactive cell is pointed with an arrowhead. Scale bar in (b) is 10 µm, applies to (b, c). In the hippocampus, the number of 29-day-old BrdU+ cells was fewer in dependent rats compared to nondependent rats, but was not different from control (d). In the mPFC, 29-day-old BrdU+ cells were greater in dependent rats compared to nondependent as well as control rats (E). n = 6, 9, and 6 for control, nondependent, and dependent rats, respectively. *p < 0.05 compared to control rats; # p < 0.05 compared to nondependent rats
Phenotypic analysis of 29-day-old BrdU labelled cells
In the hippocampal granule cell layer, the percent of 29-day-old BrdU+/NeuN+ were greater than BrdU+/GFAP+ colabeled cells, which was greater than BrdU+/NeuN-/GFAP-cells (main effect of cell-phenotype; F[2,34] = 111, p < 0.0001; Fig. 5f). No differences were revealed in phenotype of hippocampal BrdU+ cells between control, dependent and nondependent rats (interaction; F[4,34] = 1.48, p = 0.8; main effect of group F[2, 17] = 1.22, p = 0.2; Fig. 5f). In the mPFC, 29-day-old BrdU+/Olig2+ was greater than both BrdU+/GFAP+ and BrdU+/Olig2-/GFAP-cells (main effect of cell-phenotype; F[2,36] = 667, p < 0.0001; Fig. 5l), however, the latter two were not different from each other. No differences were revealed in the phenotype of mPFC BrdU+ cells between control, dependent and nondependent rats (interaction; F[4,36] = 0.713, p = 0.8; main effect of group F[2, 18] = 0.702, p = 0.3; Fig. 5l).

Alcohol dependence and nondependent drinking does not alter the phenotypic distribution of 29-day-old BrdU cells in the hippocampus and medial prefrontal cortex. (a–e, Hippocampus) Representative confocal z-stack images of 29-day-old BrdU+ cells (a, Cy3, red) colabeled with NeuN (b, FITC, green) and GFAP (c, CY5, blue). Arrowheads in a–d point to the BrdU+ cell; scale bar is 10 μm in panel (a), applies a–d. e Orthogonal view of the BrdU cell indicated in (a), shows that the cell is BrdU+/NeuN+ cell with both antibodies demonstrating equal penetration in xz and yz planes. f Quantitative analysis of BrdU-labeled cells in the hippocampus expressing NeuN or GFAP or neither in control, dependent (DEP) and nondependent (NON-DEP) rats [BrdU+/NeuN-/GFAP- (BrdU+ Only, solid bars), BrdU+/GFAP+/NeuN− (textured bars) and BrdU+/NeuN+/GFAP− (hashed bars)]. (g–k, mPFC) Representative confocal z-stack images of 29-day-old BrdU+ cells (g, Cy3, red) colabeled with Olig2 (h, FITC, green) and GFAP (i, CY5, blue). Arrowheads in g–j point to BrdU+/Olig2+ cell; arrow in H points to a Olig2+ cell that is not BrdU+ . Scale bar in G is 10 μm, applies g–j. k Orthogonal view of the BrdU cell indicated in (g), shows that the cell is BrdU+/Olig2+ cell with both antibodies demonstrating equal penetration in xz and yz planes. l Quantitative analysis of BrdU-labeled cells in the mPFC expressing Olig2 or GFAP or neither in control, dependent (DEP) and nondependent (NON-DEP) rats [BrdU+/Olig2-/GFAP- (BrdU+ Only, solid bars), BrdU+/GFAP+/Olig2- (textured bars) and BrdU+/Olig2+/GFAP− (hashed bars)]
BDNF, TrkB and phospho-TrkB expression in CIE animals
In the hippocampus, phosphorylation of TrkB receptors (pTrkB) and BDNF expression was increased during acute withdrawal (3 h post CIE) in relation to ethanol naïve control as well as protracted abstinence conditions (21d post CIE); [TrkB: F[2,30] = 13.5; p < 0.001; BDNF (the standard deviation for the protracted abstinence group was 3.6 and 2.8 fold greater than control and acute withdrawal groups (respectively) and therefore, Students t test was used to analyze hippocampal BDNF expression): (t = 3.172, p ≤ 0.01; Fig. 6b, d]. Western blotting for the total TrkB in hippocampal tissue samples yielded two bands (Fig. 6b); the top dark band was used for quantification of hippocampal total TrkB expression. No differences were revealed between control, acute withdrawal, and protracted abstinence groups in total TrkB receptor expression (tTrkB; F[2,29] = 0.880, p = 0.4 Fig. 6b, d). The ratio of pTrkB to tTrkB in the hippocampus was increased during acute withdrawal compared to control (F[2,30] = 3.72; p < 0.05; Fig. 6d), but not in relation to protracted abstinence.

BDNF and phospho-TrkB expression is increased in the hippocampus, whereas BDNF and total-TrkB expression is increased in the mPFC during acute withdrawal. a Schematic of the experimental timeline for the Western blotting analyses. Schematic for tissue punches and sample western blots from hippocampus (b) and mPFC (c). Con control, AW acute withdrawal, PA protracted abstinence. Densitometric analysis of tissue for levels of BDNF, phosphorylated-TrkB; pTrkB, total TrkB; tTrkB, and the ratio of pTrkB to tTrkB from the hippocampual (d) and mPFC (e) tissue homogenate from dependent rats following acute withdrawal (AW) and protracted abstinence (PA). Values are mean ± SEM expressed as % control, where control is represented by the dotted line in each graph. *p < 0.05, **p < 0.01, ***p < 0.001 compared to controls, # p < 0.05, ## p < 0.01 compared to AW
In the mPFC, expression of both BDNF and tTrkB were increased during acute withdrawal compared to control and protracted abstinence (F[2,32] = 7.48 and F[2,35] = 6.17, respectively; p’s < 0.01; Fig. 6c, e). In contrast, pTrkB as well as the ratio pTrk/tTrkB did not differ between control, acute withdrawal, and protracted abstinence groups (F[2,32] = 1.63, p = 0.2, and F[2,32] = 2.70, p = 0.08, respectively; Fig. 6c, e).
Discussion
Goals of this study were to examine withdrawal-induced effects on proliferation and survival of progenitors in the SGZ and mPFC and on the signaling of neurotrophic factor BDNF in rats that demonstrate both physical and motivational symptoms of alcohol dependence and compare them to nondependent animals. According to the current hypothesis, withdrawal from CIE and ethanol drinking produced a rebound effect in the number of proliferating cells which was visualized as a proliferative burst in the mPFC of dependent rats, an effect that was also observed in the hippocampal SGZ. These withdrawal induced proliferative profiles were not observed in ethanol drinking nondependent rats. Further, survival of mPFC progenitors was increased, which was contrary to the decreased survival observed in the SGZ, suggesting differential regulation of the development of progenitors with distinct mature phenotypes; i.e. neurogenesis in the hippocampus and oligodendrogenesis in the mPFC (Palmer et al. 2000; Mandyam et al. 2007b; Kim et al. 2015). Thus, protracted abstinence was associated with reduced neurogenesis of hippocampal progenitors, and with increased oligodendrogenesis of the mPFC progenitors. The current study also found that the drinking in dependent rats during the 7th week of CIE positively correlated with the proliferative burst in the SGZ observed 72 h post-CIE, but not with BAL. In contrast, the magnitude of physical withdrawal positively correlated with BALs, but not with drinking or proliferation, suggesting that the motivational effects of dependence are not mechanistically linked to level of intoxication or to physical withdrawal. Acute withdrawal from CIE enhanced BDNF expression and TrkB receptor activation in the hippocampus, and increased BDNF and TrkB expression in the mPFC, which may contribute to a conducive environment for the proliferative burst (Lee et al. 2000; Sairanen et al. 2005; Donovan et al. 2008; Cowansage et al. 2010). Finally, pro-neurotrophic microenvironment was absent during protracted abstinence, and the maladaptive progenitor survival profile in hippocampus (reduced neurons) and mPFC (increased oligodrendrocytes) was associated with the enhanced ethanol drinking even after prolonged abstinence. These results provide new insights into brain state and potential mechanisms related to alcohol relapse.
Effects in the hippocampus
The current study amalgamated previous findings that CIE induces motivational (escalated ethanol intake) and somatic (physical) signs of alcohol dependence, and withdrawal of CIE induces a reactive proliferative burst in the hippocampus (Valdez et al. 2002; O’Dell et al. 2004; Richardson et al. 2008; Hansson et al. 2010; Vendruscolo et al. 2012). These results should be interpreted with caution as BrdU may be incorporated into cells undergoing DNA repair, and because BrdU itself was found to be mitogenic in vitro [for review (Taupin 2007)], both of which may confound the magnitude of the proliferative effects reported herein. However, it is important to note that the proliferative effects of alcohol withdrawal have been confirmed with endogenous markers of cell cycle (Nixon and Crews 2004; Hansson et al. 2010). The current results of the absence of a relation between hippocampal proliferative burst and physical withdrawal were observed in the gavage model of dependence (Nixon and Crews 2004). Furthermore, the surviving cells in the hippocampal granule cell layer were primarily neuronal, consistent with the phenotype of newly born cells in the hippocampus, and suggesting that the currently used dose of BrdU did not artificially inflate cell division and survival via its mitogenic properties. A novel contribution of the current study is that ethanol drinking in dependent rats predicted the increases in cell proliferation in the hippocampus which was associated with enhanced expression of BDNF and activation of TrkB. Mechanistically, enduring changes in trophic factors may regulate cellular proliferation in the SGZ (Pencea et al. 2001; Scharfman et al. 2005; Pilar-Cuellar et al. 2013). For example, BDNF infusion into the hippocampus was shown to produce ectopic proliferation in the SGZ, hilus, and molecular layer (Scharfman et al. 2005). BDNF binds to its receptor TrkB, localized at both pre- and postsynaptic sites of glutamatergic synapses in the hippocampus; receptor binding induces receptor dimerization and phosphorylation at the autophosphorylation site Tyr-706 (Drake et al. 1999; Huang and Reichardt 2001; Chao 2003). During acute withdrawal, the enhanced BDNF levels in the hippocampus were associated with enhanced phosphorylation of TrkB at Tyr-706 without significant alterations in tTrkB expression. The lack of effect of CIE on hippocampal tTrkB is consistent with other models of alcohol administration, such as two-bottle choice exposure (Briones and Woods 2013) and liquid diet (Zhang et al. 2000). Importantly, the increased pTrkB-706 could indicate a BDNF-mediated enhanced TrkB activation, which is linked to hyperglutamatergic activity in the hippocampus (Marini et al. 1998; Perez-Gomez and Tasker 2013). Since, hippocampal neuronal activity positively and bi-directionally interacts with hippocampal neurogenesis (van Praag et al. 1999; Derrick et al. 2000; Snyder et al. 2001; Farmer et al. 2004; Chun et al. 2006, 2009), this hyperglutamatergic activity may also contribute to the early proliferative effects of CIE withdrawal (Mandyam 2013). Additionally, these proliferative effects of early withdrawal were not observed in nondependent animals, and therefore, together with the previously discussed correlational results, may suggest that the motivational aspects in alcohol dependence are predictive of reactive hippocampal proliferation.
Current results support that the hippocampal cells born during proliferative burst are not stable (Hansson et al. 2010), as evidenced by decreased survival of 29-day-old BrdU+ cells. Importantly, although cell survival was decreased in the dependent rats, alcohol self-administration with and without CIE did not alter the phenotype of these newly born progenitors compared to controls; the majority of the newly born cells continued to exhibit neuronal phenotype. Thus, suppression of survival of progenitors indicates decreased hippocampal neurogenesis. The attenuated neurogenesis in the current model contrasts the enhanced hippocampal neurogenesis observed following alcohol withdrawal using the intragastric intubation model (Nixon and Crews 2004; Nixon et al. 2008). One explanation for these conflicting results could be methodological differences, that is, differences in daily BAL levels, the duration of ethanol exposure, and the behavioral correlates associated with each paradigm (Mandyam 2013); however, an alternative explanation is also possible. Although voluntary alcohol drinking was not evaluated in the gavage model, the increase in neurogenesis was posited to contribute to the clinical observation of recovery of brain volume and function following abstinence (Nixon and Crews 2004; van Eijk et al. 2013). With CIE, the decreased progenitor survival was associated with persistence of enhanced alcohol drinking following alcohol abstinence, suggesting that the attenuated hippocampal neurogenesis may contribute to increased relapse propensity, where relapse is inferred from drinking following prolonged abstinence (Gilpin et al. 2008b; Vengeliene et al. 2014). Although these neurogenic mechanisms have not been evaluated in the clinical subjects with alcohol use disorder, absence of recovery of brain volume has been reported primarily in individuals who relapsed after a long period of abstinence (Demirakca et al. 2011; Durazzo et al. 2011), thus suggesting different translational value for both CIE and the gavage models.
Changes in hippocampal BDNF have been reported to depend upon the alcohol availability, for example continued vs intermittent access, as well as acute vs protracted withdrawal. For instance, hippocampal BDNF protein was decreased following continuously administered liquid ethanol diet (Miller and Mooney 2004), but was increased following repeated episodic administration of the same diet (Miller 2004). In CIE animals, BDNF mRNA was enhanced during acute withdrawal (12 h post-vapor cessation), an effect that was not evident during ethanol exposure (Tapia-Arancibia et al. 2001). The current report extends our understanding by showing an increase in both BDNF expression and in BDNF signaling via TrkB receptor during acute withdrawal (3 h post CIE); however, this effect was transient and did not persist into protracted abstinence (21d post CIE). Although, similar increases in BDNF during acute withdrawal and normalization during protracted abstinence have been reported in other model of alcohol exposure (Alele and Devaud 2013; Briones and Woods 2013; McClain et al. 2014), the functional implication of this transience is unclear. A recent study reported that the transient decreased BDNF/TrkB signaling corresponded with decreased hippocampal proliferation and immature neurons in the hippocampus 7 days after the cessation of chronic two-bottle choice drinking (Briones and Woods 2013). Therefore, the decreased TrkB mediated BDNF signaling following protracted abstinence could result in depletion of the hippocampal neurogenic niche (Li et al. 2008), and may reduce the survival of hippocampal progenitors born during the proliferative burst (Sairanen et al. 2005), and thereby contribute to the persistence of elevated alcohol drinking observed following CIE withdrawal (Gilpin et al. 2008b).
Effects in the medial prefrontal cortex
There is a deficit in our understanding of whether forced withdrawal from drug exposure alters proliferation, differentiation, and survival of glial progenitors in the mPFC (Cohen et al. 2014; Kim and Mandyam 2014). With respect to alcohol, continued CIE paradigm decreased proliferation and survival of progenitors in the mPFC (Richardson et al. 2009; Kim et al. 2015). In accordance with the hypothesis of the current study, this suppression of proliferation was found to be transient, and a proliferative burst was observed 72 h post CIE. Contrary to the observations in the hippocampus, the mPFC progenitors survived into protracted abstinence (29d post CIE). This is important because most of the newly born progenitors in the mPFC exhibit an oligodendroglial phenotype (Mandyam et al. 2007b; Mandyam and Koob 2012; Kim et al. 2015; Somkuwar et al. 2014), and mature into myelinating oligodendrocytes that maintain myelin plasticity (Rivers et al. 2008). One hypothesis may be that the enhanced proliferation and survival in mPFC is a compensatory (neuroprotective) mechanism that contributes to the recovery of the cortical tissue lost due to dependence (van Eijk et al. 2013; Navarro and Mandyam 2015). However, elevated drinking observed in the dependent rats following protracted abstinence does not align with this hypothesis, as recovery of brain volume was linked to reduced relapse (Demirakca et al. 2011; Durazzo et al. 2011). Notably, recent clinical evidence supports a critical role of myelination in the neuropathology of alcoholism; myelin in the PFC was decreased during protracted abstinence, which was linked to functional hyperactivity of the PFC and to stress-induced relapse (Seo et al. 2013). Although, enhanced proliferation in the mPFC did not correlate with motivational aspects of dependence in the current study, the imbalance in oligodendroglial homeostasis (proliferation and survival burst) may contribute to the persistence of alcohol seeking following protracted abstinence. Thus, whether newly generated oligodendrocyte progenitors in the mPFC of alcohol dependent rats eventually mature to attain a myelinating oligodendroglial phenotype warrants detailed investigation. Finally, withdrawal and protracted abstinence from alcohol dependence may result in maladaptive myelination in the mPFC by modulating the various signaling pathways that regulate oligodendrocyte maturation (Navarro and Mandyam 2015).
Cortical BDNF expression is differentially regulated following acute [no change (Logrip et al. 2009)], chronic episodic [no change (Miller 2004)], chronic continuous intake [increase (Miller and Mooney 2004)], and chronic escalating intake [decrease (Logrip et al. 2009)] of alcohol. Our findings add to these studies by demonstrating that acute withdrawal from CIE enhances BDNF and tTrkB expression in the mPFC, an effect that did not persist at 21d post cessation of CIE. Interestingly, the increased expression of receptor and its substrate were not associated with hyperphosphorylation of TrkB at Tyr-706, indicating that enhanced BDNF in the mPFC did not necessarily enhance TrkB signaling. One explanation for these observations may be that elevated BDNF in the mPFC alters TrkB activation in other brain regions that receive projections from mPFC; for example, PFC is a well-known source of striatal BDNF (Kokaia et al. 1993; Baquet et al. 2004). However, the lack of effect on TrkB receptor signaling in the mPFC was surprising, especially, given the role of BDNF and TrkB signaling in proliferation and survival of oligodendrocyte progenitors, and in modulation of myelination by mature oligodendroglia in several regions of the cortex [(Nakajima et al. 2010; Wong et al. 2013; Ramos-Cejudo et al. 2015; Tsiperson et al. 2015); for review, (Bankston et al. 2013)]. Therefore, the up-regulation of tTrkB receptors during acute withdrawal could be associated with a compromised physiological function of BDNF in mPFC. This may further lend credence to the hypothesis that the enhanced mPFC proliferation may be maladaptive in nature (for example, non-myelin forming) and contribute to the perpetuation of the addictive phenotype.
In conclusion, the current study reveals that withdrawal from CIE results in enhanced cell proliferation in the hippocampus and the mPFC; enhanced BDNF expression and signaling may partially underlie these changes. The enhanced cell proliferation, particularly in the hippocampal SGZ, was related to the motivational but not to the physical effects of alcohol dependence. The upregulation of BDNF signaling in the hippocampus was transient as these changes did not persist into protracted abstinence (21d post CIE), thus altering the neurogenic niche needed for the survival of the hippocampal progenitors. In contrast to the hippocampus, BDNF increase in the mPFC was not associated with TrkB activation in CIE rats, and progenitor survival was not decreased, suggesting that alternate BDNF pathways mediate survival in mPFC. These neurobiological mechanisms in the hippocampus and mPFC may contribute to the enhanced ethanol consumption that persists following prolonged abstinence from ethanol drinking . Although a minor limitation of the current study is the correlative findings, exploring potential causal link between enhanced BDNF and enhanced proliferation in the hippocampus and the mPFC in CIE animals is an interesting and important future pursuit.
References
Alele PE, Devaud LL (2013) Expression of cFos and brain-derived neurotrophic factor in cortex and hippocampus of ethanol-withdrawn male and female rats. J Pharmacol Pharmacother 4:265–274
Badanich KA, Becker HC, Woodward JJ (2011) Effects of chronic intermittent ethanol exposure on orbitofrontal and medial prefrontal cortex-dependent behaviors in mice. Behav Neurosci 125:879–891
Bankston AN, Mandler MD, Feng Y (2013) Oligodendroglia and neurotrophic factors in neurodegeneration. Neurosci Bull 29:216–228
Baquet ZC, Gorski JA, Jones KR (2004) Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci 24:4250–4258
Bechara A, Dolan S, Denburg N, Hindes A, Anderson SW, Nathan PE (2001) Decision-making deficits, linked to a dysfunctional ventromedial prefrontal cortex, revealed in alcohol and stimulant abusers. Neuropsychologia 39:376–389
Bergami M, Rimondini R, Santi S, Blum R, Gotz M, Canossa M (2008) Deletion of TrkB in adult progenitors alters newborn neuron integration into hippocampal circuits and increases anxiety-like behavior. Proc Natl Acad Sci USA 105:15570–15575
Breunig JJ, Arellano JI, Macklis JD, Rakic P (2007) Everything that glitters isn’t gold: a critical review of postnatal neural precursor analyses. Cell Stem Cell 1:612–627
Briones TL, Woods J (2013) Chronic binge-like alcohol consumption in adolescence causes depression-like symptoms possibly mediated by the effects of BDNF on neurogenesis. Neuroscience 254:324–334
Cameron HA, McKay RD (2001) Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. Journal of Comparative Neurology 435:406–417
Chao MV (2003) Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci 4:299–309
Chun SK, Sun W, Park JJ, Jung MW (2006) Enhanced proliferation of progenitor cells following long-term potentiation induction in the rat dentate gyrus. Neurobiol Learn Mem 86:322–329
Chun SK, Sun W, Jung MW (2009) LTD induction suppresses LTP-induced hippocampal adult neurogenesis. NeuroReport 20:1279–1283
Cohen A, Soleiman MT, Talia R, Koob GF, George O, Mandyam CD (2014) Extended access nicotine self-administration with periodic deprivation increases immature neurons in the hippocampus. Psychopharmacology (Berl)
Courtney KE, Polich J (2009) Binge drinking in young adults: data, definitions, and determinants. Psychol Bull 135:142–156
Cowansage KK, LeDoux JE, Monfils MH (2010) Brain-derived neurotrophic factor: a dynamic gatekeeper of neural plasticity. Curr Mole Pharmacol 3:12–29
Crews FT, Boettiger CA (2009) Impulsivity, frontal lobes and risk for addiction. Pharmacol Biochem Behav 93:237–247
Crews FT, Nixon K (2009) Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol 44:115–127
Criado JR, Liu T, Ehlers CL, Mathe AA (2011) Prolonged chronic ethanol exposure alters neuropeptide Y and corticotropin-releasing factor levels in the brain of adult Wistar rats. Pharmacol Biochem Behav 99:104–111
Dayer AG, Ford AA, Cleaver KM, Yassaee M, Cameron HA (2003) Short-term and long-term survival of new neurons in the rat dentate gyrus. J Comp Neurol 460:563–572
Demirakca T, Ende G, Kammerer N, Welzel-Marquez H, Hermann D, Heinz A, Mann K (2011) Effects of alcoholism and continued abstinence on brain volumes in both genders. Alcohol Clin Exp Res 35:1678–1685
Derrick BE, York AD, Martinez JL Jr (2000) Increased granule cell neurogenesis in the adult dentate gyrus following mossy fiber stimulation sufficient to induce long-term potentiation. Brain Res 857:300–307
Donovan MH, Yamaguchi M, Eisch AJ (2008) Dynamic expression of TrkB receptor protein on proliferating and maturing cells in the adult mouse dentate gyrus. Hippocampus 18:435–439
Drake CT, Milner TA, Patterson SL (1999) Ultrastructural localization of full-length trkB immunoreactivity in rat hippocampus suggests multiple roles in modulating activity-dependent synaptic plasticity. J Neurosci 19:8009–8026
Duque A, Rakic P (2011) Different effects of bromodeoxyuridine and [3H]thymidine incorporation into DNA on cell proliferation, position, and fate. J Neurosci 31:15205–15217
Durazzo TC, Tosun D, Buckley S, Gazdzinski S, Mon A, Fryer SL, Meyerhoff DJ (2011) Cortical Thickness, Surface Area, and Volume of the Brain Reward System in Alcohol Dependence: Relationships to Relapse and Extended Abstinence. Alcohol Clin Exp Res 35(6):1187–1200
Eadie BD, Redila VA, Christie BR (2005) Voluntary exercise alters the cytoarchitecture of the adult dentate gyrus by increasing cellular proliferation, dendritic complexity, and spine density. J Comp Neurol 486:39–47
Ernst AJ, Dempster JP, Yee R, Dennis C, Nakano L (1976) Alcohol toxicity, blood alcohol concentration and body water in young and adult rats. J Stud Alcohol 37:347–356
Farmer J, Zhao X, van Praag H, Wodtke K, Gage FH, Christie BR (2004) Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague-Dawley rats in vivo. Neuroscience 124:71–79
Galinato MH, Orio L, Mandyam CD (2015) Methamphetamine differentially affects BDNF and cell death factors in anatomically defined regions of the hippocampus. Neuroscience 286:97–108
George O, Sanders C, Freiling J, Grigoryan E, Vu S, Allen CD, Crawford E, Mandyam CD, Koob GF (2012) Recruitment of medial prefrontal cortex neurons during alcohol withdrawal predicts cognitive impairment and excessive alcohol drinking. Proc Natl Acad Sci U S A 109:18156–18161
Gilpin NW, Richardson HN, Cole M, Koob GF (2008a) Vapor inhalation of alcohol in rats. Curr Protoc Neurosci Chapter 9:Unit 9:29
Gilpin NW, Richardson HN, Lumeng L, Koob GF (2008b) Dependence-induced alcohol drinking by alcohol-preferring (P) rats and outbred Wistar rats. Alcohol Clin Exp Res 32:1688–1696
Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW (2007) Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci 10:1029–1037
Gray JD, Milner TA, McEwen BS (2013) Dynamic plasticity: the role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience 239:214–227
Hagg T (2009) From neurotransmitters to neurotrophic factors to neurogenesis. Neuroscientist 15:20–27
Hansson AC, Nixon K, Rimondini R, Damadzic R, Sommer WH, Eskay R, Crews FT, Heilig M (2010) Long-term suppression of forebrain neurogenesis and loss of neuronal progenitor cells following prolonged alcohol dependence in rats. Int J Neuropsychopharmacol 13:583–593
Holmes A, Fitzgerald PJ, MacPherson KP, DeBrouse L, Colacicco G, Flynn SM, Masneuf S, Pleil KE, Li C, Marcinkiewcz CA, Kash TL, Gunduz-Cinar O, Camp M (2012) Chronic alcohol remodels prefrontal neurons and disrupts NMDAR-mediated fear extinction encoding. Nat Neurosci 15:1359–1361
Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24:677–736
Kempermann G, Gast D, Kronenberg G, Yamaguchi M, Gage FH (2003) Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development 130:391–399
Kim A, Mandyam CD (2014) Methamphetamine affects cell proliferation in the medial prefrontal cortex: a new niche for toxicity. Pharmacol Biochem Behav 126:90–96
Kim A, Zamora-Martinez ER, Edwards S, Mandyam CD (2015) Structural reorganization of pyramidal neurons in the medial prefrontal cortex of alcohol dependent rats is associated with altered glial plasticity. Brain Struct Funct 220(3):1705–1720
Kokaia Z, Bengzon J, Metsis M, Kokaia M, Persson H, Lindvall O (1993) Coexpression of neurotrophins and their receptors in neurons of the central nervous system. Proc Natl Acad Sci USA 90:6711–6715
Kolb B, Pedersen B, Ballermann M, Gibb R, Whishaw IQ (1999) Embryonic and postnatal injections of bromodeoxyuridine produce age-dependent morphological and behavioral abnormalities. J Neurosci 19:2337–2346
Koob GF, Volkow ND (2010) Neurocircuitry of addiction. Neuropsychopharmacology 35:217–238
Kroener S, Mulholland PJ, New NN, Gass JT, Becker HC, Chandler LJ (2012) Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PLoS One 7:e37541
Kuwagata M, Ogawa T, Nagata T, Shioda S (2007) The evaluation of early embryonic neurogenesis after exposure to the genotoxic agent 5-bromo-2′-deoxyuridine in mice. Neurotoxicology 28:780–789
Lee J, Duan W, Long JM, Ingram DK, Mattson MP (2000) Dietary restriction increases the number of newly generated neural cells, and induces BDNF expression, in the dentate gyrus of rats. J Mol Neurosci 15:99–108
Li Y, Luikart BW, Birnbaum S, Chen J, Kwon CH, Kernie SG, Bassel-Duby R, Parada LF (2008) TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 59:399–412
Liu X, Weiss F (2002) Reversal of ethanol-seeking behavior by D1 and D2 antagonists in an animal model of relapse: differences in antagonist potency in previously ethanol-dependent versus nondependent rats. J Pharmacol Exp Ther 300:882–889
Logrip ML, Janak PH, Ron D (2009) Escalating ethanol intake is associated with altered corticostriatal BDNF expression. J Neurochem 109:1459–1468
Macey DJ, Schulteis G, Heinrichs SC, Koob GF (1996) Time-dependent quantifiable withdrawal from ethanol in the rat: effect of method of dependence induction. Alcohol 13:163–170
MacLennan AJ, Lee N, Walker DW (1995) Chronic ethanol administration decreases brain-derived neurotrophic factor gene expression in the rat hippocampus. Neurosci Lett 197:105–108
Majchrowicz E (1975) Induction of physical dependence upon ethanol and the associated behavioral changes in rats. Psychopharmacologia 43:245–254
Mandyam CD (2013) The interplay between the hippocampus and amygdala in regulating aberrant hippocampal neurogenesis during protracted abstinence from alcohol dependence. Frontiers in psychiatry 4:61
Mandyam CD, Koob GF (2012) The addicted brain craves new neurons: putative role for adult-born progenitors in promoting recovery. Trends Neurosci 35:250–260
Mandyam CD, Harburg GC, Eisch AJ (2007a) Determination of key aspects of precursor cell proliferation, cell cycle length and kinetics in the adult mouse subgranular zone. Neuroscience 146:108–122
Mandyam CD, Wee S, Eisch AJ, Richardson HN, Koob GF (2007b) Methamphetamine self-administration and voluntary exercise have opposing effects on medial prefrontal cortex gliogenesis. J Neurosci 27:11442–11450
Marini AM, Rabin SJ, Lipsky RH, Mocchetti I (1998) Activity-dependent release of brain-derived neurotrophic factor underlies the neuroprotective effect of N-methyl-D-aspartate. J Biol Chem 273:29394–29399
McClain JA, Morris SA, Marshall SA, Nixon K (2014) Ectopic hippocampal neurogenesis in adolescent male rats following alcohol dependence. Addict Biol 19(4):687–699
McGough NN, He DY, Logrip ML, Jeanblanc J, Phamluong K, Luong K, Kharazia V, Janak PH, Ron D (2004) RACK1 and brain-derived neurotrophic factor: a homeostatic pathway that regulates alcohol addiction. J Neurosci 24:10542–10552
Miller MW (2004) Repeated episodic exposure to ethanol affects neurotrophin content in the forebrain of the mature rat. Exp Neurol 189:173–181
Miller MW, Mooney SM (2004) Chronic exposure to ethanol alters neurotrophin content in the basal forebrain-cortex system in the mature rat: effects on autocrine-paracrine mechanisms. J Neurobiol 60:490–498
Ming GL, Song H (2005) Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci 28:223–250
Minichiello L (2009) TrkB signalling pathways in LTP and learning. Nat Rev Neurosci 10:850–860
Nakajima H, Uchida K, Yayama T, Kobayashi S, Guerrero AR, Furukawa S, Baba H (2010) Targeted retrograde gene delivery of brain-derived neurotrophic factor suppresses apoptosis of neurons and oligodendroglia after spinal cord injury in rats. Spine (Phila Pa 1976) 35:497–504
Navarro AI, Mandyam CD (2015) Protracted abstinence from chronic ethanol exposure alters the structure of neurons and expression of oligodendrocytes and myelin in the medial prefrontal cortex. Neuroscience 293:35–44
Nestler EJ (2002) Common molecular and cellular substrates of addiction and memory. Neurobiol Learn Mem 78:637–647
Nixon K, Crews FT (2004) Temporally specific burst in cell proliferation increases hippocampal neurogenesis in protracted abstinence from alcohol. J Neurosci 24:9714–9722
Nixon K, Kim DH, Potts EN, He J, Crews FT (2008) Distinct cell proliferation events during abstinence after alcohol dependence: microglia proliferation precedes neurogenesis. Neurobiol Dis 31:218–229
O’Dell LE, Roberts AJ, Smith RT, Koob GF (2004) Enhanced alcohol self-administration after intermittent versus continuous alcohol vapor exposure. Alcohol Clin Exp Res 28:1676–1682
O’Dell LE, Chen SA, Smith RT, Specio SE, Balster RL, Paterson NE, Markou A, Zorrilla EP, Koob GF (2007) Extended access to nicotine self-administration leads to dependence: circadian measures, withdrawal measures, and extinction behavior in rats. J Pharmacol Exp Ther 320:180–193
Ogawa T, Kuwagata M, Muneoka KT, Shioda S (2005) Neuropathological examination of fetal rat brain in the 5-bromo-2′-deoxyuridine-induced neurodevelopmental disorder model. Congenital anomalies 45:14–20
Oscar-Berman M, Marinkovic K (2003) Alcoholism and the brain: an overview. Alcohol Res Health 27:125–133
Palmer TD, Willhoite AR, Gage FH (2000) Vascular niche for adult hippocampal neurogenesis. J Comp Neurol 425:479–494
Paxinos G, Watson C (1997) The rat brain in stereotaxic coordinates, 3°, edition edn. Academic Press, San Diego
Pencea V, Bingaman KD, Wiegand SJ, Luskin MB (2001) Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J Neurosci 21:6706–6717
Perez-Gomez A, Tasker RA (2013) Transient domoic acid excitotoxicity increases BDNF expression and activates both MEK- and PKA-dependent neurogenesis in organotypic hippocampal slices. BMC Neurosci 14:72
Pian JP, Criado JR, Milner R, Ehlers CL (2010) N-methyl-D-aspartate receptor subunit expression in adult and adolescent brain following chronic ethanol exposure. Neuroscience 170:645–654
Pilar-Cuellar F, Vidal R, Diaz A, Castro E, dos Anjos S, Pascual-Brazo J, Linge R, Vargas V, Blanco H, Martinez-Villayandre B, Pazos A, Valdizan EM (2013) Neural plasticity and proliferation in the generation of antidepressant effects: hippocampal implication. Neural Plast 2013:537265
Rajkowska G, Miguel-Hidalgo JJ (2007) Gliogenesis and glial pathology in depression. CNS Neurol Disord: Drug Targets 6:219–233
Ramos-Cejudo J, Gutierrez-Fernandez M, Otero-Ortega L, Rodriguez-Frutos B, Fuentes B, Vallejo-Cremades MT, Hernanz TN, Cerdan S, Diez-Tejedor E (2015) Brain-derived neurotrophic factor administration mediated oligodendrocyte differentiation and myelin formation in subcortical ischemic stroke. Stroke 46:221–228
Richardson HN, Lee SY, O’Dell LE, Koob GF, Rivier CL (2008) Alcohol self-administration acutely stimulates the hypothalamic-pituitary-adrenal axis, but alcohol dependence leads to a dampened neuroendocrine state. Eur J Neurosci 28:1641–1653
Richardson HN, Chan SH, Crawford EF, Lee YK, Funk CK, Koob GF, Mandyam CD (2009) Permanent impairment of birth and survival of cortical and hippocampal proliferating cells following excessive drinking during alcohol dependence. Neurobiol Dis 36:1–10
Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD (2008) PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci 11:1392–1401
Rosenbloom M, Sullivan EV, Pfefferbaum A (2003) Using magnetic resonance imaging and diffusion tensor imaging to assess brain damage in alcoholics. Alcohol Res Health 27:146–152
Rowell JJ, Ragsdale CW (2012) BrdU birth dating can produce errors in cell fate specification in chick brain development. J Histochem Cytochem 60:801–810
Russo SJ, Mazei-Robison MS, Ables JL, Nestler EJ (2009) Neurotrophic factors and structural plasticity in addiction. Neuropharmacology 56(Suppl 1):73–82
Sairanen M, Lucas G, Ernfors P, Castren M, Castren E (2005) Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J Neurosci 25:1089–1094
SAMHSA (2013) Results from the 2012 National Survey on Drug Use and Health: Summary of National Findings,. NSDUH Series H-46 HHS Publication No. (SMA) 13–4795
Scharfman H, Goodman J, Macleod A, Phani S, Antonelli C, Croll S (2005) Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp Neurol 192:348–356
Sekerkova G, Ilijic E, Mugnaini E (2004) Bromodeoxyuridine administered during neurogenesis of the projection neurons causes cerebellar defects in rat. J Comp Neurol 470:221–239
Seo D, Lacadie CM, Tuit K, Hong KI, Constable RT, Sinha R (2013) Disrupted ventromedial prefrontal function, alcohol craving, and subsequent relapse risk. JAMA Psychiatry 70:727–739
Snyder JS, Kee N, Wojtowicz JM (2001) Effects of adult neurogenesis on synaptic plasticity in the rat dentate gyrus. J Neurophysiol 85:2423–2431
Somkuwar SS, Staples MC, Galinato MH, Fannon MJ, Mandyam CD (2014) Role of NG2 expressing cells in addiction: a new approach for an old problem. Frontiers in pharmacology 5:279
Staples MC, Kim A, Mandyam CD (2015) Dendritic remodeling of hippocampal neurons is associated with altered NMDA receptor expression in alcohol dependent rats. Mol Cell Neurosci 65:153–162
Stephens DN, Duka T (2008) Review. Cognitive and emotional consequences of binge drinking: role of amygdala and prefrontal cortex. Philos Trans R Soc Lond B Biol Sci 363:3169–3179
Sullivan EV, Rosenbloom MJ, Pfefferbaum A (2000a) Pattern of motor and cognitive deficits in detoxified alcoholic men. Alcohol Clin Exp Res 24:611–621
Sullivan EV, Rosenbloom MJ, Lim KO, Pfefferbaum A (2000b) Longitudinal changes in cognition, gait, and balance in abstinent and relapsed alcoholic men: relationships to changes in brain structure. Neuropsychology 14:178–188
Tapia-Arancibia L, Rage F, Givalois L, Dingeon P, Arancibia S, Beauge F (2001) Effects of alcohol on brain-derived neurotrophic factor mRNA expression in discrete regions of the rat hippocampus and hypothalamus. J Neurosci Res 63:200–208
Taupin P (2007) BrdU immunohistochemistry for studying adult neurogenesis: paradigms, pitfalls, limitations, and validation. Brain Res Rev 53:198–214
Tsiperson V, Huang Y, Bagayogo I, Song Y, VonDran MW, DiCicco-Bloom E, Dreyfus CF (2015) Brain-derived neurotrophic factor deficiency restricts proliferation of oligodendrocyte progenitors following cuprizone-induced demyelination. ASN neuro 7(1). doi:10.1177/1759091414566878
Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP, Koob GF (2002) Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res 26:1494–1501
van Eijk J, Demirakca T, Frischknecht U, Hermann D, Mann K, Ende G (2013) Rapid partial regeneration of brain volume during the first 14 days of abstinence from alcohol. Alcohol Clin Exp Res 37:67–74
van Praag H, Christie BR, Sejnowski TJ, Gage FH (1999) Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci USA 96:13427–13431
Vendruscolo LF, Barbier E, Schlosburg JE, Misra KK, Whitfield TW Jr, Logrip ML, Rivier C, Repunte-Canonigo V, Zorrilla EP, Sanna PP, Heilig M, Koob GF (2012) Corticosteroid-dependent plasticity mediates compulsive alcohol drinking in rats. J Neurosci 32:7563–7571
Vengeliene V, Bilbao A, Spanagel R (2014) The alcohol deprivation effect model for studying relapse behavior: a comparison between rats and mice. Alcohol 48:313–320
Welinder C, Ekblad L (2011) Coomassie staining as loading control in Western blot analysis. J Proteome Res 10:1416–1419
Wong AW, Xiao J, Kemper D, Kilpatrick TJ, Murray SS (2013) Oligodendroglial expression of TrkB independently regulates myelination and progenitor cell proliferation. J Neurosci 33:4947–4957
Zhang L, Dhillon HS, Barron S, Hicks RR, Prasad RM, Seroogy KB (2000) Effects of chronic ethanol administration on expression of BDNF and trkB mRNAs in rat hippocampus after experimental brain injury. Brain Res Mol Brain Res 79:174–179
Acknowledgments
The study was supported by funds from the National Institute on Drug Abuse (DA022473 and DA034140 to CDM), National Institute on Alcoholism and Alcohol Abuse (NIAAA; AA020098 and AA06420 to CDM) and Alcohol Beverage Medical Research Foundation (to CDM), a NIAAA Pathway to Independence Award to SE (LSUHSC-New Orleans, AA020839), and T32AA00747 to MCS. We appreciate the technical support of Robert Lintz, Ilham Polis, and Maury Cole for assistance with operant self-administration studies and ethanol vapor chambers. We thank Atoosa Ghofranian from the independent study program at UCSD for assistance with tissue processing. We thank Drs. Charles Stiles and John Alberta, Harvard Medical School, for generously providing the Olig2 antibody. We thank Dr. Marian Lorgrip for critical reading of the manuscript. This is manuscript number 27067 from The Scripps Research Institute. The authors have no conflicts of interest to report.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Somkuwar, S.S., Fannon, M.J., Staples, M.C. et al. Alcohol dependence-induced regulation of the proliferation and survival of adult brain progenitors is associated with altered BDNF-TrkB signaling. Brain Struct Funct 221, 4319–4335 (2016). https://doi.org/10.1007/s00429-015-1163-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00429-015-1163-z