
Abstract
Alzheimer’s disease (AD) is an untreatable neurodegenerative disease that deteriorates memory. Increased physical/cognitive activity reduces dementia risk by promoting neuronal and glial response. Although few studies have investigated microglial response in wild-type rodents following exposure to physical/cognitive stimulation, environmental-induced changes of microglia response to AD have been neglected. We investigated effects of running (RUN) and enriched (ENR) environments on numerical density (N v, #/mm3) and morphology of microglia in a triple transgenic (3×Tg-AD) mouse model of AD that closely mimics AD pathology in humans. We used immunohistochemical approach to characterise microglial domain by measuring their overall cell surface, volume and somata volume. 3×Tg-AD mice housed in standard control (STD) environment showed significant increase in microglial N v (11.7 %) in CA1 stratum lacunosum moleculare (S.Mol) of the hippocampus at 12 months compared to non-transgenic (non-Tg) animals. Exposure to combined RUN and ENR environments prevented an increase in microglial N v in 3×Tg-AD and reduced microglial numbers to non-Tg control levels. Interestingly, 3×Tg-AD mice housed solely in ENR environment displayed significant decrease in microglial N v in CA1 subfield (9.3 % decrease), stratum oriens (11.5 % decrease) and S.Mol (7.6 % decrease) of the hippocampus compared to 3×Tg-AD mice housed in STD environment. Morphological analysis revealed microglial hypertrophy due to pronounced increase in microglia surface, volume and somata volume (61, 78 and 41 %) in 3×Tg-AD mice housed in RUN (but not in ENR) compared to STD environment. These results indicate that exposure to RUN and ENR environments have differential effects on microglial density and activation-associated changes in microglial morphology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative pathology that depreciates cognitive functions due to specific damage of brain areas involved in learning and memory including neocortex and the hippocampus (Braak and Braak 1991). Neuropathological features of AD brains include extracellular deposition of beta amyloid (Aβ) in the form of senile/neuritic plaques, intracellular neurofibrillary tangles (NFTs), synaptic loss and neuronal cell death (Selkoe 2002). Extracellular Aβ deposition triggers glial activation that is a common neuropathological feature of AD brains (Beach et al. 1989).
Originated from myelomonocytes, microglia are the resident immune-cells that enter the developing central nervous system (CNS) and undergo phenotypic metamorphosis to resting microglia, which detect and respond to both physiological and pathological changes in adult CNS homeostasis (van Rossum and Hanisch 2004; Kettenmann et al. 2011, 2013; Ransohoff and Perry 2009). In the healthy brain microglia are usually found in a “resting” state characterised by small somata and fine processes that continuously monitor their surroundings (Nimmerjahn et al. 2005). Pathology-induced alterations in CNS homeostasis trigger microglia remodelling, generally referred to microglial activation. Morphologically, this activation process is characterised by larger cell bodies and small/thicker processes (Kettenmann et al. 2011).
Regular physical activity and higher education level act as protective factors in preventing cognitive decline and reducing the risk of developing AD in the elderly population (Laurin et al. 2001; Wang et al. 2002). Exposure to voluntary wheel running (RUN) and enriched environment (ENR) in rodents had been designed to mimic the physical and cognitive “active” lifestyle in humans (Cracchiolo et al. 2007; Wood et al. 2010). These environments have profound effects on adult brain, which reacts to the increased physical activity and elevated complexity in the environmental condition. Exposure to RUN and ENR environments have been associated with improved memory performance and increased adult neurogenesis (Nilsson et al. 1999; Kempermann et al. 2002). In addition to their effect on hippocampal neurogenesis, exposure to RUN and ENR also induce profound effects on microglia (Steiner et al. 2004; Ehninger and Kempermann 2003; Ehninger et al. 2011). Although the early study by Altman and Das (1964) reported glial proliferation in rat cortex following exposure to ENR (Altman and Das 1964), more recent studies suggest region-specific alterations in microglia following exposure to RUN and ENR housing conditions (Ehninger and Kempermann 2003; Ehninger et al. 2011; Ziv et al. 2006).
Adult mice housed in RUN for 10 days showed an increase in the number of newly produced microglia (measured using Iba1 and BrdU staining) in the superficial layers of the cortex (Ehninger and Kempermann 2003). Similarly, exposure to ENR for 6 weeks triggered an increase in the number of resident isolectin B4 (IB-4)-positive microglia and newly produced microglia (measured using IB-4 and BrdU staining) in the rat hippocampus (Ziv et al. 2006). In contrast, more recent findings have shown that exposure to RUN (10 days) and ENR (40 days) reduced new-born microglia number in the amygdala of adult mice (Ehninger et al. 2011), suggesting regions-specificity of microglial alterations upon housing in RUN and ENR environments.
Environmental-induced changes in microglia using animal models of AD have been, hitherto, almost completely neglected. A single study in PS1ΔE9 and PS1M146L mouse models of AD revealed no changes in Iba1-positive microglia population in the hippocampus following exposure to ENR (1 h per day for 1 month) (Choi et al. 2008). The PS1ΔE9 and PS1M146L mouse models of AD, however, exhibit only partial AD-related neuropathology. Given the absence of studies in other mouse models of AD, environmental-induced changes in microglia response requires further clarification. In addition, no studies have addressed quantitative changes in microglia morphology following exposure to RUN and ENR in adult brains.
In the present study, we have investigated the effect of chronic (9 months) exposure to RUN and ENR on the numerical density (N v, #/mm3) and overall morphology of hippocampal microglia in the triple transgenic (3×Tg-AD) mouse model of AD, which expresses both Aβ plaques and NFTs with a spatiotemporal distribution that closely resembling AD pathology in humans; this model also exhibits age-related cognition deficits (Clinton et al. 2007; Oddo et al. 2003a, b; Frazer et al. 2008).
Materials and methods
All animal procedures were carried out in accordance with the United Kingdom Animals (Scientific Procedures) Act of 1986 under the license from the Home Office and were approved by the Animal Care and Ethical Committee at The University of Manchester (PCD 50/2506). All efforts were made to reduce the number of animals by following the 3Rs (reduction, refinement and replacement).
Animals and experimental design
The procedure for generating the triple transgenic (3×Tg-AD) mouse model of AD has been described in details previously (Oddo et al. 2003a, b; Rodríguez et al. 2008, 2009). Briefly, human cDNA harbouring the Swedish APP mutation (KM670/671NL) and human P301L four repeats mutated tau harbouring the P301L (tauP301L) mutations (both under control of the mouse Thy1.2 regulatory element) were co-microinjected into a single-cell embryo of a homozygous PS1M146V knock-in mouse (Oddo et al. 2003a, b). The background of the PS1 knock-in mice is a hybrid 129/C57BL6. The non-transgenic (non-Tg) control mice used were from the same strain and genetic background as the PS1 knock-in mice, but they express the endogenous wild-type mouse PS1 gene. All 3×Tg-AD and non-Tg control mice were obtained by crossing homozygous breeders. The animals were housed in the same-sex cage, kept in 12 h light–dark cycles with free access to food and water.
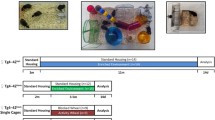
Following birth and weaning period (P21), the animals were housed in groups of at least four animals in standard laboratory environment until 3 months of age (Lafenetre et al. 2010; Rodríguez et al. 2011). Both 3×Tg-AD and non-Tg control mice were randomly assigned into control (STD, n = 5 for 3×Tg-AD and n = 4 non-Tg) housing, voluntary wheel running (RUN, n = 5 for 3×Tg-AD, n = 4 for non-Tg) or environmental enriched (ENR, n = 5 for both 3×Tg-AD and non-Tg) for period of 9 months (Fig. 1a) as previously considered and described (Catlow et al. 2009; Rodríguez et al. 2011; Wolf et al. 2006). The RUN group had free and unlimited access to a running wheel (Fig. 1b). The ENR housing was composed of coloured tunnels, climbing materials and a tilted running wheel (Fig. 1c). These mice were kept in the enrichment cages with the environment being regularly rearranged to maintain novelty (Cracchiolo et al. 2007), whilst the STD groups were kept in their respective standard cages. The animals were kept in STD, RUN and ENR environment for a period of 9 months, till they reached 12 months of age. We had chosen 12 months of age because our previous studies have shown that 3×Tg-AD animals displayed increase in both resting and activated microglial population in the hippocampus at this age (Rodríguez et al. 2010). In addition, we studied exclusively male mice to avoid the confounding effects of oestrous in female animals.

Experimental design. Following the weaning period (P21) male 3×Tg-AD and non-transgenic (Non-Tg) animals were housed in standard laboratory (STD) housing environment until 3 months of age. At 3 months of age animals were randomly assigned into either voluntary wheel running (RUN), environmental enriched (ENR) or STD condition for a period of 6 months a. The photographs show the set-up for RUN environment with a voluntary running wheel b and ENR environment containing coloured tunnels, climbing materials and tilted running wheels c. All animals were sacrificed by perfusion at 12 months of age. CTR standard housing cage, RUN large cage with unlimited access to a running wheel, ENR enriched cage
Fixation and tissue processing
Male 3×Tg-AD and their respective non-Tg control mice were anaesthetised with intraperitoneal injection of sodium pentobarbital. Mice were perfused through the aortic arch with 3.75 % acrolien (TAAB, UK) in a solution of 2 % paraformaldehyde (PFA, Sigma, UK) and 0.1 M phosphate buffer (PB) pH 7.4, followed by 2 % PFA. Brains were then removed and cut into 4–5 mm coronal slabs of tissue and were post-fixed in 2 % PFA for 24 h. Coronal sections of the brain were cut into 40–50 μm thick slices using a vibrating microtome (Leica, UK). Free floating brain sections were stored in cryoprotectant solution containing 25 % sucrose and 3.5 % glycerol in 0.05 M PB at pH 7.4. Coronal hemisections at levels −1.70 mm/−2.46 mm (hippocampus) posterior to bregma were selected for immunohistochemistry according to the mouse brain atlas of Paxinos and Franklin (2004).
Antibodies
A rabbit antiserum raised against the synthetic peptide corresponding to the C-terminus of ionised calcium binding adaptor 1 (Iba1, Wako, Germany) was used to identify microglial cells. To confirm that Iba1 antibody labels all microglial population in the hippocampus, we used Iba1 immunohistochemistry in the dorsal hippocampal section of CX3CR1+/eGFP mice, which selectively express Enhanced Green Fluorescent Protein in microglial cells (Fig. 2) (Jung et al. 2000; Srivastava et al. 2005). A polyclonal affinity-purified rat antiserum raised against CD11b (Mac-1, Serotec, UK) was used for the determination of activated microglial cells (Solovjov et al. 2005; Rodríguez et al. 2010).

Confocal micrographs showing the dorsal hippocampus of CX3CR1+/eGFP mice (a, d), Iba1-immunoreactive microglia (b, e) and their respective colocalisation (c, f). Note that Iba1-positive microglia stains all microglia population in the dorsal hippocampus. Scale bars a–c = 200 μm, d–f = 20 μm
Finally, for the identification of Aβ plaques a monoclonal mouse antibody against the amino acid residues 1–16 of Aβ (Covance, USA) was used. The specificity of these antibodies has been reported previously using immunohistochemistry (Noristani et al. 2010; Rodríguez et al. 2010; Olabarria et al. 2011; Yoshikawa et al. 2010) and western blot analysis (Yang et al. 2010). To assess for non-specific background labelling or cross reactivity between antibodies derived from different host species, a series of control experiments were performed. Omission of primary and/or secondary antibodies from the incubation solutions resulted in a total absence of target labelling.
Immunohistochemistry
Brain sections were incubated for 30 min in 30 % methanol and 3 % hydrogen peroxide (H2O2, Sigma, UK) in 0.1 M PB followed by 0.1 M PB (5 min) and 1 % sodium borohydride (Aldrich, UK, 30 min). Sections were washed with 0.1 M PB profusely and placed in 0.1 M trizma base saline (TS, 10 min) followed by incubation in 0.5 % albumin bovine serum (BSA, Sigma, UK) in 0.1 M TS and 0.25 % Triton (Sigma, UK) for 30 min and 48 h incubation in rabbit anti-Iba1 (1:2,000) primary antibody. The sections were rinsed in 0.1 M TS for 30 min and incubated in 1:200 dilution of Alexa 488 goat anti-rabbit (Invitrogen, UK) IgG, secondary antibody for 1 h at followed by rinsing in 0.1 M TS.
For the dual peroxidase labelling sections were incubated for 48 h in both primary antibodies cocktail (1) rabbit anti-Iba1 (1:2,000) and (2) mouse anti-beta amyloid monoclonal antibody (1:2,000) simultaneously. The Iba1 and Aβ antibodies were detected in a sequential manner on the same sections. For Iba1 labelling, the sections were incubated in 1:200 dilutions of biotinylated donkey anti-rabbit IgG (Jackson Immunoresearch, UK) for 1 h followed by incubation for 30 min in avidin–biotin peroxidase complex (ABC, Vector Laboratories Ltd., UK). The peroxidase reaction product was visualised by incubation in a solution containing 0.022 % of 3,3′diaminobenzidine (Aldrich, UK) and 0.003 % H2O2. For Aβ labelling, the same brains sections were incubated in 1:200 dilution of biotinylated horse anti-mouse IgG followed by incubation for 30 min in ABC. The Aβ peroxidase reaction was visualised by incubation in SGZ kit (Vector Laboratories Ltd., UK) as described previously (Rodríguez et al. 2008, 2011). This allowed us to see the Iba1 labelling in brown, thus differentiating it from the Aβ neuritic plaques labelled in blue as they were visualised and revealed by a SGZ kit.
For triple immunofluorescence labelling, the sections were incubated for 48 h at room temperature in primary antibody cocktail (1) rabbit anti-Iba1 (1:2,000), (2) mouse anti-Aβ (1:2,000) and (3) rat anti-Mac-1 (1:1,000) simultaneously. Subsequently, each antigen was detected in a sequential manner on the same sections by incubation with Alexa 595 goat anti-mouse, Alexa 488 goat anti-rabbit and Alexa 633 goat anti-rat (Invitrogen, UK). Finally, sections were rinsed with 0.1 M PB for 30 min and permanently mounted in an aqueous medium.
Quantification of Iba1 positive microglia
The numerical density (N v, #/mm3) of Iba1-positive microglia were analysed in the CA1 subfield of the hippocampus. We used five representative non-consecutive sections throughout the dorsal hippocampus (analysed in an area of 235,000 μm2 for CA1 in coronal sections of 40 μm thickness). To ensure unbiased microglial counting, representative sections were selected in random, taken into account that they represented similar anatomical level of the hippocampus and its individual layers. The specific analysed areas were the stratum oriens (S.Or), the stratum radiatum (S.Rad) and the stratum lacunosum moleculare (S.Mol) of the hippocampal CA1 subfield. Confocal stack images were used for this propose. Hippocampal boundaries were clearly delineated, thus, counts were reproducible. Iba1-positive microglia cells were intensely labelled against dark background that made them easy to identify with equal chance of being counted. All counting of Iba1-positive microglia cells were conducted by a single investigator (H.N.N) who was blind to the mice genotype and housing environments, therefore, counting bias was kept to a minimum.
Optical density (OD) measurement
Using computer-assisted imaging analysis (ImageJ 1.32j, NIH, Bethesda, MD, USA), we analysed the density of intraneuronal Aβ by measuring their optical density (OD), as we have described previously (Noristani et al. 2010). In brief and to exclude any experimental errors and/or bias, all images were taken at a constant light intensity. The OD was calculated from a relative scale of intensity ranging from 0 to 255, with a measurement of 255 corresponding to the area with very low intraneuronal Aβ accumulation and 0 corresponding to the densest area of labelling (Noristani et al. 2010). The calibration density was kept constant for measuring all section to avoid experimental variances. Non-specific OD in sections was measured from the corpus callosum. The density of intraneuronal Aβ was measured in stratum pyramidale (PCL), of CA1, CA2 and CA3 subfields of the hippocampus in the 3×Tg-AD mouse model of AD at 12 months of age following chronic exposure to STD, RUN and ENR housing. To analyse the changes in intraneuronal Aβ against constant control, 255 was divided by the control region (corpus callosum) and the obtained factor was multiplied by the region of interest in every given section (Noristani et al. 2010). Inverse optical density was obtained by subtracting from the obtained background level (255). Measurement of mean density were taken and averaged, after background subtraction, from each hippocampal layers in both the left and the right hemisphere of each slice. The results are shown as inverse optical density (IOD/pixel).
Morphological analysis of microglial domain
Microglia (n = 30–35) were imaged using confocal scanning microscopy (Olympus, inverted), recording layers at every 0.6 μm. Parallel confocal planes were superimposed and morphological analysis was carried out by cell analyst programme (Chvatal et al. 2007) using digital filters (average 3 × 3, convolution, gauss 5 × 5, despeckle, simple objects removal) to determine the surface (S) and the volume (V) of Iba1-immunostained microglia, as previously described (Olabarria et al. 2010; Yeh et al. 2011).
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). Unpaired t tests were used to examine differences in the N v, surface and volume of Iba1 labelled microglia between the 3×Tg-AD and non-Tg control animals in different housing environment. Significance was accepted at p ≤ 0.05. The data were analysed using GraphPad Prism 4.0 (GraphPad Software, Inc.).
Results
Heterogeneous distribution of microglial cells in different layers of hippocampus
In the CA1 subfield of both non-Tg control and 3×Tg-AD mice the density of Iba1-positive microglia varied between hippocampal layers (Fig. 3). The highest density of Iba1-positive microglia was found in the stratum lacunosum moleculare (S. Mol) of CA1 subfield compared to other hippocampal strata (on average 11,363 in 3×Tg-AD and 10,777 Iba1-positive cells/mm3 in non-Tg control, Fig. 3a–f). The stratum oriens (S.Or) and the stratum radiatum (S.Rad) have less microglia (on average 8,011 in 3×Tg-AD and 8,457 Iba1-positive cells/mm3 in non-Tg control within S.Or and 8,025 in 3×Tg-AD and 8,120 Iba1-positive cells/mm3 in non-Tg control within S.Rad), whereas Iba1-positive microglia was rarely observed in the stratum pyramidale of the hippocampus. This difference in distribution of microglia within different hippocampal layers was observed irrespective of genotype and housing environment (Fig. 3).

Confocal micrographs showing Iba1-immunoreactive microglia in the dorsal hippocampus of non-Tg (a–c) and 3×Tg-AD (d–f) mice housed in STD (a, d), RUN (b, e) and ENR (c, f) environments. Scale bars a–f = 100 μm, a′–f′ = 50 μm
Morphologically, we classified microglial cells into two groups: (i) resting microglia displayed small cell bodies equipped with thin to-medium ramified processes extending to the surrounding neuropil, and (ii) activated microglia characterised by enlarged cell bodies with short and thick processes (Fig. 3a–f).
3×Tg-AD mice in STD environment displayed increased numerical density of microglia in CA1 S. Mol
Global analysis of CA1 subfield (all stratas combined) showed no general changes in Iba1-positive microglia cells in the CA1 subfield of the hippocampus between 3×Tg-AD and non-Tg control mice housed in either in STD, RUN and ENR environments (p = 0.3085, Fig. 4a). However, a detailed quantitative analysis of individual CA1 layers revealed that 3×Tg-AD mice housed in STD condition displayed increased N v of Iba1-positive microglia compared to non-Tg group in the CA1 S.Mol (11.7 % increase, p = 0.0183, Fig. 4d), which also exhibited the highest density of microglial density in both 3×Tg-AD and non-Tg control groups. No changes were observed in microglial density between 3×Tg-AD and non-Tg control housed in RUN and ENR environments (Fig. 4d). Similarly, CA1 S.Or and CA1 S.Rad showed no changes in microglial density between 3×Tg-AD and non-Tg control mice housed in STD, RUN or ENR housing environments (p = 0.5503 for S.Or, p = 0.9062 for S.Rad, Fig. 4b, respectively).

Bar graphs showing the effect of RUN and ENR environment on the N v of Iba1-positive microglia in the different strata of the CA1 subfield of hippocampus a. Bars represent mean ± SEM, *p < 0.05, **p < 0.01 compared to 3×Tg-AD animals in the STD housing environment, # p < 0.05 compared to non-Tg in the same housing environment
ENR environment decreased microglial numerical density in CA1 region in 3×Tg-AD mice
3×Tg-AD mice housed in ENR environment showed significantly reduced numerical density of Iba1-positive microglia in the CA1 subfield of the hippocampus compared to 3×Tg-AD group housed in STD housing environment (9.3 % decrease, p = 0.0077, Fig. 4a). Analysis of individual CA1 strata revealed that ENR-induced decrease in Iba1-positive microglia was significant in the CA1 S.Or (11.5 % decrease, p = 0.0162, Fig. 4b) and CA1 S.Mol (7.6 % decrease, p = 0.0335, Fig. 4d), but not in CA1 S.Rad (7.3 % decrease, p = 0.1131, Fig. 4c). No environmental-induced changes were observed in non-Tg control group neither in CA1 subfield nor in other hippocampal strata (Fig. 4).
RUN environment potentiated microglial activation in 3×Tg-AD animals
To determine whether environmental stimulation affects microglial morphology we analysed the surface (μm2), volume (μm3) and somata volume (μm3) of Iba1-positive microglia in 3×Tg-AD mice exposed to STD, RUN and ENR housing environments (Fig. 5). Since no changes were observed in the N v of microglia in non-Tg control animals, the latter group was not included in morphological analysis. Iba1-positive microglia in 3×Tg-AD mice exposed to RUN environment showed a significant increase in surface (61 %, p = 0.0217, Fig. 5b, d) and volume (78 %, p = 0.0104, Fig. 5b, e) when compared to 3×Tg-AD animals in STD environment. RUN-induced increase in microglia surface and volume was also accompanied by a significant increase in somata volume (41 %, p = 0.0098, Fig. 5b, f). No significant differences were observed in microglial surface, volume and somata volume in 3×Tg-AD animals exposed to ENR environment when compared to STD group.

Confocal micrographs showing environmental-induced changes in microglial morphology in 3×Tg-AD mice housed in STD a, RUN b and ENR c environments. Scale bars a–c = 20 μm. Bar graphs showing environmental-induced changes in microglial surface d, volume e and somata volume f in the 3×Tg-AD mouse model of AD. Bars represent mean ± SEM, *p < 0.05, **p < 0.01 compared to 3×Tg-AD animals in the STD housing environment
Environmental-induced alterations in microglia is independent of Aβ neuropathology in 3×Tg-AD animals
To examine whether RUN and ENR-induced alteration in microglia is driven by Aβ neuropathology, we studied Aβ neuropathology in 3×Tg-AD mice housed in RUN and ENR compared to that of STD housing at 12 months of age. Our immunohistochemical analysis showed pronounced intraneuronal Aβ deposition with rate Aβ neuritic plaques at 12 months of age in 3×Tg-AD mice housed in RUN, ENR and STD environments. Our quantitative analysis of intraneuronal Aβ density showed no changes in 3×Tg-AD animals upon exposure to RUN or ENR compared to that STD housing condition (supplementary Fig. 1).
Increased Aβ deposition was clearly evident at 18-month-old 3×Tg-AD mice. Dual peroxidase labelling revealed high density of Aβ neuritic plaques in the CA1 subfield of the hippocampus that was mainly surrounded by activated Iba1-positive microglia in the 3×Tg-AD mouse model of AD at 18 months of age (Fig. 6a, b). Iba1-positive microglial cells located distal to Aβ plaques, showed typical ramified features including small cell body with large/thin ramified processes (Fig. 6c).

Brightfield micrographs showing Iba1 and Aβ immunoreactivity in the dorsal hippocampus of 3×Tg-AD mice a with both amoeboid b and ramified c Iba1-positive microglia. Scale bars a = 100 μm, b, c = 20 μm
Triple immunofluorescence labelling revealed high density of Iba1 and Mac-1 positive microglia, adjacent to Aβ plaques and Aβ aggregates. In particular, triple labelling showed Aβ aggregate engulfment inside activated microglial somata that was characterised by Iba1 and Mac-1 staining (Fig. 7).

Confocal micrographs showing amoeboid microglia characterised by Iba1 a and MAC-1 b labelling that is actively taking up Aβ c in microglia somata. Scale bars a–d = 10 μm
Discussion
The major finding from the current study is that 3×Tg-AD mouse model of AD, housed in standard (STD) housing environment, displays strata-specific increase in microglial numerical density (N v, #/mm3) in CA1 S.Mol that was reversed following chronic exposure to running (RUN) and enriched (ENR) housing environments. In particular, RUN and ENR significantly reduced microglia N v in 3×Tg-AD mice compared to their counterparts housed in STD environment. Furthermore, 3×Tg-AD mice house in RUN, but not ENR, displayed pronounced microglial hypertrophy compared to 3×Tg-AD mice housed in STD environment. Interestingly, intraneuronal Aβ density remained stable in both RUN and ENR housing conditions suggesting that environmental-induced changes in microglia are independent of amyloid pathology in 3×Tg-AD mouse model of the disease.
Increased microglial density in the 3×Tg-AD mouse model of AD
Our finding of increased microglial N v in CA1 S.Mol of 3×Tg-AD mice housed in STD environment (Fig. 4d), is in agreement with our previous report where we found an increased density of both ramified and activated microglia in the CA1 subfield of the hippocampus at 12 months of age (Rodríguez et al. 2010). Other studies, including our recent findings, suggest that increased microglia density in 3×Tg-AD mice closely resembles the evolution of AD-related neuropathology with the earliest increase in microglial density occurring in the entorhinal cortex [6 months, (Janelsins et al. 2005)] followed by the CA1 subfield [9–12 months, (Rodríguez et al. 2013)] and the dentate gyrus [DG, 18 months, (Rodríguez et al. 2013)] of the hippocampus. Post-mortem AD brains also showed a specific spatiotemporal evolution of AD-related neuropathology, with the initial degenerative signs appearing in entorhinal cortex that is then spread towards the hippocampus and other cortical areas (Van Hoesen and Hyman 1990; Braak and Braak 1991; Ohm 2007).
AD-associated increase in microglia density has been consistently reported in post-mortem AD brains (McGeer et al. 1987) and in numerous transgenic mouse models of AD with excessive Aβ neuropathology namely: APPswe (Frautschy et al. 1998), PS1×APP, (Jimenez et al. 2008) and APPswe/PS1d9×YFP mice (Meyer-Luehmann et al. 2008).
Although the triggering mechanism for AD-related increase in microglial density is not currently known, it is likely to be due to amyloidogenic accumulation of not only Aβ neuritic plaques but also smaller Aβ aggregates (Rodríguez et al. 2010; Matsuoka et al. 2001). Previous studies have shown that hippocampal Aβ plaque formation is initiated between 9 and 12 months and becomes more evident at 18 months and older age in 3×Tg-AD mice (Oddo et al. 2003a, b; Rodríguez et al. 2008). The initiation of extracellular Aβ aggregates in 3×Tg-AD mice directly correlates with increased microglial density in this brain region (Rodríguez et al. 2010).
Activated microglia accumulates around Aβ plaques. Dual peroxidase labelling revealed that Aβ plaques are surrounded by Iba1-positive microglia that display different morphological features depending on their vicinity to Aβ plaques (Fig. 6). Specifically, microglia located adjacent to Aβ plaques displayed activated status as revealed by an increase in their soma and appearance of thicker processes (Fig. 6b), whilst microglia located at a distance from Aβ plaques showed typical ramified features including small soma with thin/medium ramified processes (Fig. 6c). Triple florescence labelling revealed that activated microglia (characterised by Iba1 and Mac-1 immunostaining) are actively involved in the uptake of Aβ aggregate inside microglial somata (Fig. 7). Given the close association between Aβ aggregation and increased microglial density, it is likely that Aβ deposition may act as an underlying mechanism for stimulating increased microglial density in 3×Tg-AD mice.
Exposure to ENR environment reduces microglia population in the 3×Tg-AD mouse model of AD independent of Aβ neuropathology
The finding of ENR-induced decrease in hippocampal microglia population (Fig. 4) is contradictory to a previous report in PS1ΔE9 and PS1M146L mice, which showed no changes in Iba1-positive hippocampal microglia following exposure to ENR (Choi et al. 2008). Multiple factors may account for contradictory findings including (i) experimental designs, (ii) transgenic line and (iii) the age of the transgenic mice.
Specifically, Choi et al. (2008) exposed PS1ΔE9 and PS1M146L mice in ENR 1 h per day for a period of 1 month as opposed to the current study where the 3×Tg-AD mice were continuously housed in ENR for 9 months (see “Materials and methods”). In addition, PS1ΔE9 and PS1M146L mice only express Aβ plaques, with no evident NFT neuropathology as opposed to 3×Tg-AD mice, which expressed both Aβ plaques and NFTs that closely resembles AD evolution in humans (Oddo et al. 2003a, b). Furthermore, Choi et al. (2008) investigated environmental-induced alterations in microglia proliferation in young (2 months) mice compared to 12 months of age in the current study. As mentioned previously, we had chosen this age because our previous report had shown increased density of both ramified and activated microglia in 3×Tg-AD mice at 12 months (Rodríguez et al. 2010). Given the age-specific increase in microglial density in different brain regions in the 3×Tg-AD mice (discussed above), differences in examined age may significantly contribute to the contradictory findings between the two studies.
Although, to our knowledge, there are no other reports on environmental-induced alterations in microglia population using transgenic models of AD, studies in wild-type mice and rats suggest region-specific changes in microglia following exposure to RUN and ENR housing (Ehninger and Kempermann 2003; Ehninger et al. 2011; Ziv et al. 2006). Exposure to RUN and ENR increased microglial proliferation in the mice cortex (Ehninger and Kempermann 2003) and the rat hippocampus (Ziv et al. 2006), while reducing the number of new-born microglia in the mice amygdala (Ehninger et al. 2011).
Unlike ENR, exposure to RUN had no significant effect in reducing hippocampal microglial N v in 3×Tg-AD mice (Fig. 4). Such ENR-induced decrease in microglial N v may be due to the “complete” enriched environment that includes not only increased physical activity but also increased social/cognitive stimulation compared to that of RUN (Cracchiolo et al. 2007). Each of the above factors may have a synergistic effect on microglial N v over and above achieved by increased physical activity in RUN environment (van Praag et al. 2000).
Exposure to RUN environment potentiates activation of microglia in the 3×Tg-AD mouse model of AD
The increase microglial hypertrophy following exposure to RUN environment is, to our knowledge, the first quantitative report on environmental-induced alteration on microglial morphology in a transgenic mouse model of AD (Fig. 5). Similarly, although previous studies have reported both increased and reduced microglial proliferation in rodents following exposure to RUN and ENR environments (Ehninger and Kempermann 2003; Ehninger et al. 2011; Ziv et al. 2006), no study has investigated microglia hypertrophy via quantitative analysis of microglia morphology. Interestingly, Ehninger and Kempermann (2003) reported no qualitative morphological changes in cortical microglia of adult mice following exposure to RUN environment (Ehninger and Kempermann 2003).
RUN-induced alterations in microglia morphology in 3×Tg-AD mice suggest that these glia cells plastically react to physiological stimuli such as exposure to RUN and ENR environments. Although the functional significance of RUN-dependent microglia hypertrophy remains ambiguous, previous reports have shown close association between increased microglial activation and adult neurogenesis (Ziv et al. 2006).
Previous studies in our laboratory have reported impaired adult neurogenesis in 3×Tg-AD mice both in the dentate gyrus (Rodríguez et al. 2008) and the subventricular zone (Rodríguez et al. 2009), which was reversed following exposure to RUN and ENR environment (Rodríguez et al. 2011). Given the fact that activation of microglia promote neurogenesis both in vitro (Butovsky et al. 2006) and in vivo (Ziv et al. 2006), our results suggest that RUN-induced increased in microglia activation may participate in re-storing the impaired neurogenesis in the 3×Tg-AD mouse model of AD (Rodríguez et al. 2011).
RUN and ENR environments have no effect on Aβ pathology in 3×Tg-AD animals
Neither RUN nor ENR altered intraneuronal Aβ deposition compared to that of the STD housing in 3×Tg-AD mice (supplementary Fig. 1). These finding suggest that environmental-induced changes in microglia is independent of amyloid pathology in 3×Tg-AD mice. These findings are in agreement with Arendash et al. (2004) who also reported no changes in brain Aβ deposition in aged APPswe transgenic mice following chronic (4 months) exposure to RUN and ENR environments (Arendash et al. 2004). In addition, Wolf et al. (2006) showed stable Aβ plaque load using three different techniques (area fraction fractionator, surface area and absolute number of Aβ plaques) in the cortex and the hippocampus of 16-month-old APP23 mice upon chronic (11 months) exposure to RUN and ENR housing environment (Wolf et al. 2006). Finally, these findings are supported by Mirochnic et al. (2009) who also showed that neither RUN nor ENR housing altered the fraction of the hippocampus covered by Aβ plaques at 18 months in APP23 transgenic mice (Mirochnic et al. 2009).
Contrary, others have shown that both RUN and ENR reduces Aβ load in multiple transgenic mouse models of AD including APPswe/PS1 (Cracchiolo et al. 2007) (decreased Aβ deposition), PS1/PdAPP (Costa et al. 2007) (showed 50 % less brain beta-amyloid) and TgCRND8 transgenic mice (Adlard et al. 2005). As mentioned above, these contradictory findings may be due to differences in the experimental designs, transgenic line and the examined age of the transgenic mice.
In summary, our results indicate that RUN and ENR environment have differential effects on microglia cell density/and morphology. Specifically, whilst exposure to ENR environment reduced microglia N v, increased physical activity induced microglia hypertrophy as evident by increase in microglia surface, volume and somata volume. Such microglia activation in the 3×Tg-AD mouse model of AD highlights their involvement not only due to disease-related state, but also following physiological stimuli.
References
Adlard PA, Perreau VM, Pop V, Cotman CW (2005) Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J Neurosci 25(17):4217–4221. doi:10.1523/JNEUROSCI.0496-05.2005
Altman J, Das GD (1964) Autoradiographic examination of the effects of enriched environment on the rate of glial multiplication in the adult rat brain. Nature 204:1161–1163
Arendash GW, Garcia MF, Costa DA, Cracchiolo JR, Wefes IM, Potter H (2004) Environmental enrichment improves cognition in aged Alzheimer’s transgenic mice despite stable beta-amyloid deposition. NeuroReport 15(11):1751–1754
Beach TG, Walker R, McGeer EG (1989) Patterns of gliosis in Alzheimer’s disease and aging cerebrum. Glia 2(6):420–436. doi:10.1002/glia.440020605
Braak H, Braak E (1991) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 82(4):239–259
Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M (2006) Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci 31(1):149–160. doi:10.1016/j.mcn.2005.10.006
Catlow BJ, Rowe AR, Clearwater CR, Mamcarz M, Arendash GW, Sanchez-Ramos J (2009) Effects of environmental enrichment and physical activity on neurogenesis in transgenic PS1/APP mice. Brain Res 1256:173–179. doi:10.1016/j.brainres.2008.12.028
Choi SH, Veeraraghavalu K, Lazarov O, Marler S, Ransohoff RM, Ramirez JM, Sisodia SS (2008) Non-cell-autonomous effects of presenilin 1 variants on enrichment-mediated hippocampal progenitor cell proliferation and differentiation. Neuron 59(4):568–580. doi:10.1016/j.neuron.2008.07.033
Chvatal A, Anderova M, Hock M, Prajerova I, Neprasova H, Chvatal V, Kirchhoff F, Sykova E (2007) Three-dimensional confocal morphometry reveals structural changes in astrocyte morphology in situ. J Neurosci Res 85(2):260–271. doi:10.1002/jnr.21113
Clinton LK, Billings LM, Green KN, Caccamo A, Ngo J, Oddo S, McGaugh JL, LaFerla FM (2007) Age-dependent sexual dimorphism in cognition and stress response in the 3×Tg-AD mice. Neurobiol Dis 28(1):76–82. doi:10.1016/j.nbd.2007.06.013
Costa DA, Cracchiolo JR, Bachstetter AD, Hughes TF, Bales KR, Paul SM, Mervis RF, Arendash GW, Potter H (2007) Enrichment improves cognition in AD mice by amyloid-related and unrelated mechanisms. Neurobiol Aging 28(6):831–844. doi:10.1016/j.neurobiolaging.2006.04.009
Cracchiolo JR, Mori T, Nazian SJ, Tan J, Potter H, Arendash GW (2007) Enhanced cognitive activity—over and above social or physical activity—is required to protect Alzheimer’s mice against cognitive impairment, reduce Abeta deposition, and increase synaptic immunoreactivity. Neurobiol Learn Mem 88(3):277–294. doi:10.1016/j.nlm.2007.07.007
Ehninger D, Kempermann G (2003) Regional effects of wheel running and environmental enrichment on cell genesis and microglia proliferation in the adult murine neocortex. Cereb Cortex 13(8):845–851
Ehninger D, Wang LP, Klempin F, Romer B, Kettenmann H, Kempermann G (2011) Enriched environment and physical activity reduce microglia and influence the fate of NG2 cells in the amygdala of adult mice. Cell Tissue Res 345(1):69–86. doi:10.1007/s00441-011-1200-z
Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152(1):307–317
Frazer ME, Hughes JE, Mastrangelo MA, Tibbens JL, Federoff HJ, Bowers WJ (2008) Reduced pathology and improved behavioral performance in Alzheimer’s disease mice vaccinated with HSV amplicons expressing amyloid-beta and interleukin-4. Mol Ther 16(5):845–853. doi:10.1038/mt.2008.39
Janelsins MC, Mastrangelo MA, Oddo S, LaFerla FM, Federoff HJ, Bowers WJ (2005) Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein-1 expression specifically within the entorhinal cortex of triple transgenic Alzheimer’s disease mice. J Neuroinflamm 2:23. doi:10.1186/1742-2094-2-23
Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J (2008) Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci 28(45):11650–11661. doi:10.1523/JNEUROSCI.3024-08.2008
Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20(11):4106–4114
Kempermann G, Gast D, Gage FH (2002) Neuroplasticity in old age: sustained fivefold induction of hippocampal neurogenesis by long-term environmental enrichment. Ann Neurol 52(2):135–143. doi:10.1002/ana.10262
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91(2):461–553. doi:10.1152/physrev.00011.2010
Kettenmann H, Kirchhoff F, Verkhratsky A (2013) Microglia: new roles for the synaptic stripper. Neuron 77(1):10–18. doi:10.1016/j.neuron.2012.12.023
Lafenetre P, Leske O, Ma-Hogemeie Z, Haghikia A, Bichler Z, Wahle P, Heumann R (2010) Exercise can rescue recognition memory impairment in a model with reduced adult hippocampal neurogenesis. Front Behav Neurosci 3:34. doi:10.3389/neuro.08.034.2009
Laurin D, Verreault R, Lindsay J, MacPherson K, Rockwood K (2001) Physical activity and risk of cognitive impairment and dementia in elderly persons. Arch Neurol 58(3):498–504 (pii:noc00302)
Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O’Banion MK, Tenner AJ, Lemere CA, Duff K (2001) Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer’s disease. Am J Pathol 158(4):1345–1354. doi:10.1016/S0002-9440(10)64085-0
McGeer PL, Itagaki S, Tago H, McGeer EG (1987) Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 79(1–2):195–200
Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT (2008) Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 451(7179):720–724. doi:10.1038/nature06616
Mirochnic S, Wolf S, Staufenbiel M, Kempermann G (2009) Age effects on the regulation of adult hippocampal neurogenesis by physical activity and environmental enrichment in the APP23 mouse model of Alzheimer disease. Hippocampus 19(10):1008–1018. doi:10.1002/hipo.20560
Nilsson M, Perfilieva E, Johansson U, Orwar O, Eriksson PS (1999) Enriched environment increases neurogenesis in the adult rat dentate gyrus and improves spatial memory. J Neurobiol 39(4):569–578. doi:10.1002/(SICI)1097-4695(19990615)39
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308(5726):1314–1318. doi:10.1126/science.1110647
Noristani HN, Olabarria M, Verkhratsky A, Rodríguez JJ (2010) Serotonin fibre sprouting and increase in serotonin transporter immunoreactivity in the CA1 area of hippocampus in a triple transgenic mouse model of Alzheimer’s disease. Eur J Neurosci 32(1):71–79. doi:10.1111/j.1460-9568.2010.07274.x
Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM (2003a) Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging 24(8):1063–1070 (pii:S0197458003002033)
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003b) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39(3):409–421 (pii:S0896627303004343)
Ohm TG (2007) The dentate gyrus in Alzheimer’s disease. Prog Brain Res 163:723–740. doi:10.1016/S0079-6123(07)63039-8
Olabarria M, Noristani HN, Verkhratsky A, Rodríguez JJ (2010) Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58(7):831–838. doi:10.1002/glia.20967
Olabarria M, Noristani HN, Verkhratsky A, Rodríguez JJ (2011) Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: mechanism for deficient glutamatergic transmission? Mol Neurodegener 6:55. doi:10.1186/1750-1326-6-55
Paxinos G, Franklin KBJ (2004) The mouse brain in stereotaxic coordinates, 2nd edn. Elesvier Academic Press, San Diego
Ransohoff RM, Perry VH (2009) Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 27:119–145. doi:10.1146/annurev.immunol.021908.132528
Rodríguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, LaFerla FM, Oddo S, Verkhratsky A (2008) Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS ONE 3(8):e2935. doi:10.1371/journal.pone.0002935
Rodríguez JJ, Jones VC, Verkhratsky A (2009) Impaired cell proliferation in the subventricular zone in an Alzheimer’s disease model. NeuroReport 20(10):907–912. doi:10.1097/WNR.0b013e32832be77d
Rodríguez JJ, Witton J, Olabarria M, Noristani HN, Verkhratsky A (2010) Increase in the density of resting microglia precedes neuritic plaque formation and microglial activation in a transgenic model of Alzheimer’s disease. Cell Death Dis 1:e1. doi:10.1038/cddis.2009.2
Rodríguez JJ, Noristani HN, Olabarria M, Fletcher J, Somerville TD, Yeh CY, Verkhratsky A (2011) Voluntary running and environmental enrichment restores impaired hippocampal neurogenesis in a triple transgenic mouse model of Alzheimer’s disease. Curr Alzheimer Res 8(7):707–717 (pii:BSP/CAR/0150)
Rodríguez JJ, Noristani HN, Hilditch T, Olabarria M, Yeh CY, Witton J, Verkhratsky A (2013) Increased densities of resting and activated microglia in the dentate gyrus follow senile plaque formation in the CA1 subfield of the hippocampus in the triple transgenic model of Alzheimer’s disease. Neurosci Lett 552:129–134. doi:10.1016/j.neulet.2013.06.036
Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science 298(5594):789–791. doi:10.1126/science.1074069.298/5594/789
Solovjov DA, Pluskota E, Plow EF (2005) Distinct roles for the alpha and beta subunits in the functions of integrin alphaMbeta2. J Biol Chem 280(2):1336–1345. doi:10.1074/jbc.M406968200
Srivastava M, Jung S, Wilhelm J, Fink L, Buhling F, Welte T, Bohle RM, Seeger W, Lohmeyer J, Maus UA (2005) The inflammatory versus constitutive trafficking of mononuclear phagocytes into the alveolar space of mice is associated with drastic changes in their gene expression profiles. J Immunol 175(3):1884–1893 (pii:175/3/1884)
Steiner B, Kronenberg G, Jessberger S, Brandt MD, Reuter K, Kempermann G (2004) Differential regulation of gliogenesis in the context of adult hippocampal neurogenesis in mice. Glia 46(1):41–52. doi:10.1002/glia.10337
Van Hoesen GW, Hyman BT (1990) Hippocampal formation: anatomy and the patterns of pathology in Alzheimer’s disease. Prog Brain Res 83:445–457
van Praag H, Kempermann G, Gage FH (2000) Neural consequences of environmental enrichment. Nat Rev Neurosci 1(3):191–198. doi:10.1038/35044558
van Rossum D, Hanisch UK (2004) Microglia. Metab Brain Dis 19(3–4):393–411
Wang HX, Karp A, Winblad B, Fratiglioni L (2002) Late-life engagement in social and leisure activities is associated with a decreased risk of dementia: a longitudinal study from the Kungsholmen project. Am J Epidemiol 155(12):1081–1087
Wolf SA, Kronenberg G, Lehmann K, Blankenship A, Overall R, Staufenbiel M, Kempermann G (2006) Cognitive and physical activity differently modulate disease progression in the amyloid precursor protein (APP)-23 model of Alzheimer’s disease. Biol Psychiatry 60(12):1314–1323. doi:10.1016/j.biopsych.2006.04.004
Wood NI, Carta V, Milde S, Skillings EA, McAllister CJ, Ang YL, Duguid A, Wijesuriya N, Afzal SM, Fernandes JX, Leong TW, Morton AJ (2010) Responses to environmental enrichment differ with sex and genotype in a transgenic mouse model of Huntington’s disease. PLoS ONE 5(2):e9077. doi:10.1371/journal.pone.0009077
Yang X, He G, Hao Y, Chen C, Li M, Wang Y, Zhang G, Yu Z (2010) The role of the JAK2-STAT3 pathway in pro-inflammatory responses of EMF-stimulated N9 microglial cells. J Neuroinflamm 7:54. doi:10.1186/1742-2094-7-54
Yeh CY, Vadhwana B, Verkhratsky A, Rodriguez JJ (2011) Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer’s disease. ASN Neuro 3(5):271–279. doi:10.1042/AN20110025
Yoshikawa F, Banno Y, Otani Y, Yamaguchi Y, Nagakura-Takagi Y, Morita N, Sato Y, Saruta C, Nishibe H, Sadakata T, Shinoda Y, Hayashi K, Mishima Y, Baba H, Furuichi T (2010) Phospholipase D family member 4, a transmembrane glycoprotein with no phospholipase D activity, expression in spleen and early postnatal microglia. PLoS ONE 5(11):e13932. doi:10.1371/journal.pone.0013932
Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M (2006) Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci 9(2):268–275. doi:10.1038/nn1629
Acknowledgments
The present study was supported by Alzheimer’s Research Trust Programme Grant (ART/PG2004A/1) to JJR and AV. Grant Agency of the Czech Republic (GACR 309/09/1696 and GACR 304/11/0184) to JJR and (GACR 305/08/1381; GACR 305/08/1384) to AV. The Spanish Government, Plan Nacional de I + D + I 2008-2011 and ISCIII-Subdirección General de Evaluación y Fomento de la investigación (PI10/02738) to JJR and AV and the Government of the Basque Country grant (AE-2010-1-28; AEGV10/16) to JJR, and by the Welcome Trust.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material

{kind=link}
Rights and permissions
About this article
Cite this article
Rodríguez, J.J., Noristani, H.N. & Verkhratsky, A. Microglial response to Alzheimer’s disease is differentially modulated by voluntary wheel running and enriched environments. Brain Struct Funct 220, 941–953 (2015). https://doi.org/10.1007/s00429-013-0693-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00429-013-0693-5