
Abstract
The proper organization and function of GABAergic interneuron networks is essential for many cognitive processes and abnormalities in these systems have been documented in schizophrenic patients. The memory function of the hippocampus depends on two major patterns of oscillations in the theta and gamma ranges, both requiring the intact functioning of the network of fast-firing interneurons expressing parvalbumin. We examined the ability of acute and chronic administration of NMDA receptor (NMDA-R) antagonists to recapitulate the oscillatory dysfunctions observed in schizophrenia. In freely moving rats, acute injection of MK801 or ketamine increased gamma power in both CA1 and dentate gyrus of the hippocampus. Theta peak shifted to higher frequencies whereas the average 5–10 Hz theta power decreased by 24% in CA1 and remained high in the dentate gyrus. Strong increase in CA1 gamma and decrease in theta power triggered by brainstem stimulation were found under urethane anesthesia. In contrast to acute experiments, chronic administration of ketamine caused a steady decline in both gamma and theta oscillations, 2–4 weeks after treatment. A further important difference between the two models was that the effects of acute injection were more robust than the changes after chronic treatment. Chronic administration of ketamine also leads to decrease in the number of detectable parvalbumin interneurons. Histological examination of interindividual differences indicated, however, that within the ketamine treated group a further decrease in parvalbumin neurons correlated with strengthening of oscillations. The findings are consistent with abnormalities of oscillations in human schizophrenia and further validate the NMDA-R hypofunction hypothesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The recent shift in the conceptualization of schizophrenia from errors in dopamine neurotransmission to core deficits in information processing gave rise to a new generation of animal models focusing on the role of other transmitter systems, such as NMDA and GABA (Goff and Coyle 2001; Marcotte et al. 2001; Kilts 2002; Moghaddam and Jackson 2003). Importantly, these models exhibit the animal equivalents of schizophrenia-related neurocognitive deficits (Young et al. 2009) and show characteristic abnormalities in the organization of the GABAergic networks (Keilhoff et al. 2004; Rujescu et al. 2006; Braun et al. 2007; Morrow et al. 2007; Xi et al. 2009) reminiscent of those in schizophrenic patients (Reynolds et al. 2004; Lewis et al. 2005). GABAergic interneurons are involved in the generation of brain oscillations (Buzsaki et al. 1992; Csicsvari et al. 1999; Klausberger et al. 2003) which in turn are known to be critical for cognitive processes (Buzsaki and Draguhn 2004; Wang 2010). Alterations in brain oscillations of schizophrenic patients were proposed to significantly contribute to the neurocognitive impairments characteristic for this disease (Lewis et al. 2005; Gonzalez-Burgos and Lewis 2008).
The NMDA receptor (NMDA-R) hypofunction model of schizophrenia is primarily based on observations that acute NMDA-R blockade recapitulates, more accurately than previous dopamine-based models, schizophrenia symptoms, including cognitive deficits (Javitt and Zukin 1990; Adler et al. 1999; Kantrowitz and Javitt 2010). The validity of this model in regard to oscillatory network activity is less clear, however, as administration of ketamine to human subjects increased gamma power (Plourde et al. 1997; Hong et al. 2010) whereas most studies in schizophrenia patients reported reduced power of oscillations (Spencer et al. 2009).
Several lines of evidence support the involvement of hippocampal pathology in schizophrenia. Hippocampus-dependent cognitive functions are severely impaired in schizophrenics (Tamminga et al. 2010), NMDA-R is reduced in the hippocampus (Gao et al. 2000), and increased metabolic activity in temporal lobe (Medoff et al. 2001) indicates that NMDA-R hypofunction primarily affects GABA inhibition in the hippocampus (Lisman et al. 2008). The memory function of hippocampus depends on two major patterns of oscillations, in the theta and gamma ranges, both requiring the intact functioning of the network of fast firing interneurons, expressing parvalbumin (PV) (Buzsaki et al. 1992; Csicsvari et al. 1999; Klausberger et al. 2003). Reduced expression of GAD67 and PV in hippocampus is a consistent postmortem finding in human schizophrenics (Benes et al. 1998; Todtenkopf and Benes 1998; Zhang and Reynolds 2002) and in animal models (Berretta et al. 2004), including those based on chronic treatment with NMDA antagonists (Keilhoff et al. 2004).
The goal of this study was to test the effect of NMDA-R antagonists on the capacity of hippocampal networks to generate oscillations. In the first set of experiments, we reproduced earlier findings of NMDA-R antagonist induced acute gamma increase in the neocortex (Pinault 2008; Hakami et al. 2009), but focused our attention on parallel changes in the hippocampus and extended the analysis to include slow rhythms in the 5–10 Hz range, normally associated with active states. In the second experiment, hippocampal oscillations were investigated in a chronic rodent model of schizophrenia induced by NMDA-R blockade in which both PV cell loss (Keilhoff et al. 2004) and cognitive deficits have been demonstrated earlier (Becker et al. 2003).
Methods
All experiments followed the recommendations of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center. The experiments were performed on adult male Sprague-Dawley rats (Charles River Laboratories, Massachusetts, USA). The rats were kept in a controlled environment, on a 12 h light/dark schedule with a constant temperature of 21 ± 1°C. Food and water were available ad libitum.
Surgery
The rats were anesthetized either by a mixture of ketamine and xylazine, for all freely moving recordings (60–80 and 10 mg/kg of body weight, injected itraperitoneally), or by urethane, for experiments under anesthesia (≤1.6 g/kg body weight, intraperitoneal injections). The depth of the anesthesia was regularly controlled by pinching a hind paw to verify the lack of withdrawal reflexes, and the body temperature (37°C) was maintained using an isothermal pad. The rats were implanted with EEG screws over the frontal (AP: +1.0, L: 2.0) and parietal (AP: −2.5, L: 2.5) cortices and with pairs of 125 μm stainless steel wires with 1.0 mm tip separation to record hippocampal field potentials. One of these electrodes was positioned at or below the hippocampal fissure (AP: −3.7, L:2.2, DV: −3.5 mm relative to bregma), and the other in or above the CA1 pyramidal layer in order to pick up signals from the two major theta dipoles. Theta oscillations in these locations are in opposite phase of each other, which allows verification of correct electrode placement during the experiment. For simplicity, the oscillations generated in the CA1 pyramidal layer will be referred to as CA1-theta and those recorded in deep layers below the fissure will be referred to as DG-theta, thorough the manuscript. Two additional screws were implanted in the nasal bone and over the cerebellum to serve as ground and reference. Rats used for freely moving recordings also received two EMG electrodes in their neck muscle on either side. The electrodes were connected to a miniature connector and secured on the skull using dental cement (Hajos et al. 2008; Lanre-Amos and Kocsis 2010).
Anesthetized rats
In eight rats, anesthesia was maintained by urethane (≤1.6 g/kg body weight, intraperitoneal injections) for the entire duration of the experiment. In addition to recording electrodes, in these rats, a double cannula with 3 mm separation was implanted over the pontine reticular formation on both sides (AP: −7.8, L; ±1.5, DV: 6.0–7.0). After the recordings started, pairs of twisted wires were inserted through each cannula so their tips protruded by 1 mm. The wires were connected to a stimulator and were positioned in the reticular nucleus pontis oralis (RPO) at a depth where hippocampal theta could be elicited by the smallest stimulus intensity (10 s trains of 0.2 ms square waves at 100 Hz). The threshold to elicit theta (0.1–0.3 mA) and the maximum intensity (0.8–1.0 mA), above which theta frequency or amplitude no longer increased, was identified individually in each rat and the stimulus intensity versus theta power trend was measured before MK801 injection and for a sufficient time afterward to see the recovery of the effects. The EEG signals were filtered between 1 and 100 Hz and sampled at 1 kHz. Autospectra were computed using FFT for 4 s segments during each stimulation and the average theta power was calculated between 5 and 10 Hz and average gamma power in the 30–50 Hz range. The total power of the signals over a 10 min long segment without stimulation was used for normalization between experiments to compensate for voltage differences due to possible variations in electrode placement in hippocampal layers in individual rats. Student’s t test was used to compare theta power before versus after injection at four levels of stimulus intensity and the slopes of the linear trend between the two parameters.
Freely moving rats, acute injections
Seven to 10 days after surgery, the rats were placed in individual recording chambers and connected to the amplifier through slip ring commutators (PlasticsOne, Inc., Roanoke, USA). The recordings started at 8–10 am and lasted for 10–24 h. 4 h after the start of the recording, either ketamine (10 mg/kg, n = 6) or MK801 (0.2 mg/kg, n = 9) was injected subcutaneously. The signals were filtered between 0.3 and 100 Hz and sampled at 250 Hz. Autospectra were calculated using FFT for consecutive 16 s segments. Theta and gamma powers were calculated for each of these segments in the 5–10 and 30–50 Hz frequency range, respectively, and averaged over 5 min segments to represent the time course of changes in oscillation power. Theta and gamma power of the signals were then normalized using average values over the first hour of the recording at the day of the injection to compensate for voltage differences due to possible variations in electrode placement and tissue–electrode contact in individual rats. For statistical analysis, theta and gamma powers were averaged for 4 h before injection and for 1 and 4 h after injection for ketamine and MK801, respectively. Statistical analysis was based on two-way and one-way ANOVA, to compare the effects of drugs (ketamine and MK801) and electrode location (CA1 recorded in all rats, DG in 6 rats injected with ketamine and 4 with MK801).
Seven rats were used to compare the effects of ketamine injected subcutaneously (10 mg/kg) and intraperitoneally (10, 20, 40, 60 mg/kg). Each rat received one subcutaneous and a maximum of three intraperitoneal injections separated by at least 3 days. Frontal cortex EEG was recorded in all seven rats, CA1 electrodes were verified (using theta phase) in six, DG electrodes in four rats. The first 45 min of the recording (mostly awake motor activity) and the first 45 min after ketamine administration (which was also awake motor activity) and their ratio was used for statistical comparison between different doses and routes of administration (ANOVA with post hoc Bonferroni pair-wise comparisons).
Chronic ketamine-treated rats
Treatment with subanesthetic doses of ketamine followed the protocol described in (Becker et al. 2003; Becker and Grecksch 2004). Adult rats were implanted with EEG electrodes and allowed to recover for 1 week after surgery. Then, after 2 weeks of control recordings, the rats received 30 mg/kg ketamine (n = 11) or saline (n = 5) daily, for five consecutive days, intraperitoneally. EEG and EMG was recorded in a subset of rats (n = 5) on the first day of ketamine injection to test acute reaction. Regular schedule of recordings returned in all 16 rats 2–3 days after treatment and continued for 2–4 weeks. Six recording sessions, each 3-h long, were used for statistical analysis, three during the control period and three after the treatment, i.e. on day 2–3, 5–7, and 14–15 after the last ketamine injection. The location of the hippocampal electrode could be reliably identified in the CA1 in six and in DG in another six rats. EEG signals from frontal and parietal cortex and from hippocampus were processed as described above and autospectra were generated for 3-h long recordings. Theta and gamma power was calculated between 5–10 and 30–50 Hz, respectively and normalized using the averages of the three pre-injection recordings, i.e. expressed as percent change relative to pre-treatment control. Theta power was also calculated selectively for theta segments during waking and rapid eye movement (REM) sleep. For this analysis, all theta segments were identified using the theta/delta ratio (>4), and wake and REM sleep were differentiated using the root-mean-square value of the EMG. Statistical analysis was based on two-way ANOVA with treatment and either time or area as main factors, and one-way ANOVA to compare theta and gamma power at the 6 time points within different groups.
Histology
After the completion of the electrophysiological recordings, the rats were deeply anesthetized with urethane (2.8 mg/kg body weight) and perfused transcardially with 0.9% saline, followed by 400 ml of 4% paraformaldehyde in 0.1 M phosphate buffered saline (PBS) at pH 7.4. After overnight postfixation, the whole brains were cryoprotected in a solution of 30% sucrose in 0.1 M PBS at pH 7.4. Serial coronal sections with 40 μm thickness containing the hippocampus were cut on a freezing microtome and collected in PBS.
Free-floating brain sections were incubated sequentially with: (1) a primary mouse antibody directed against parvalbumin (Millipore) at 1:2,000 dilution in 0.1 M phosphate-buffered saline (PB) with 0.3% Triton X-100 for 2 nights at 4°C, (2) a secondary, biotinylated donkey anti-mouse antibody (1:300 dilution in PBS; Jackson Immunoresearch, West Grove, PA, USA) for 2 h, and (3) ABC Elite Kit (1:500; Vector Laboratories, Burlingame, CA, USA) for 2 h. Then the sections were processed with a mixture of diaminobenzidine (DAB; 0.2%), 0.05% hydrogen peroxide, and 0.6% nickel ammonium sulfate in 50 mM Tris buffer (pH 7.6).
After all immunostaining procedures, coronal brain sections were rinsed in PBS, mounted serially, dehydrated, and then coverslipped with Permount. To aid in the anatomical localization of the hippocampal regions (CA1, CA2, CA3, dentate gyrus), one series of coronal sections from each animal was counterstained with thionin before dehydration. The nomenclature used to describe various subregions of the hippocampus was followed according to Paxinos and Watson (1998).
Cell counting was carried out using the same procedure for brains of three saline and six ketamine treated rats, without knowledge of which experimental or control group the rat belonged to. Due to the shrinkage in the Z-plane following dehydration, stereological methods were not employed. Instead, the counting regions of the dorsal hippocampus were determined from AP −2.3 to −4.5 mm, relative to Bregma. PVB-positive neurons were counted CA1 and CA3, in all layers. Magnification was 10× objective lens and 10× ocular. 16 sections were counted for each animal. The “number” of PVB neurons refers to the total number of neurons sampled in the dorsal hippocampus from equally spaced sections.
Results
Acute effect of NMDA-R antagonists on hippocampal oscillations
In the first experiment, the effect of NMDA-R antagonists was investigated in urethane-anesthetized rats, in which hippocampal oscillations can be experimentally manipulated. High frequency stimulation of the brainstem reticular formation in this preparation induces an active state in the hippocampus characterized by strong theta rhythm (Fig. 1a). The frequency and power of this rhythm increase in response to raising the stimulus intensity, following a linear trend (Fig. 1c, pre-injection control) (Kocsis and Li 2004). MK801 had strong effects on both theta and gamma oscillations in the hippocampus, but the changes were in opposite directions (Fig. 1b). Theta power decreased after injection for all levels of stimulation (Fig. 1c) but the largest suppression occurred at high stimulus intensities (theta power 48 + 0.09% of pre-injection level; t[7] = −3.40, P = 0.011). This effectively eliminated the stimulus intensity-dependent trend in the evoked theta power, as indicated by the significant (t[7] = −3.55, P = 0.009), approximately tenfold decrease in the slope of the regression line. In contrast, gamma power increased by 58–67% in the hippocampus (paired t test, pre vs. post-injection comparison; t[7] = 2.54, P = 0.0038) and showed a strong tendency of increase in the overlying parietal cortex, although the increase in the latter was not significant (t[7] = −2.19, P = 0.065, alpha = 0.47). Surprisingly, gamma power was not affected by MK801 in the frontal cortex under urethane anesthesia (paired t test; P > 0.05).

Effect of NMDA-R antagonist on theta and gamma oscillations elicited in hippocampus and neocortex by stimulation of the brainstem reticular formation in urethane anesthetized rats. a, b Specimen recording of hippocampal EEG filtered between 4–10 Hz (hθ) and 30–50 Hz (hγ) and of EEG (30–50 Hz) recorded over the frontal (fγ) and parietal (pγ) corticies before (a) and after (b) injection of MK801 (0.2 mg/kg, s/c) along with a time marker of stimulation (10 s). c Average hippocampal theta power (4–10 Hz) elicited by electrical stimulation of the pontine reticular formation at current intensities increasing from threshold to maximum (see “Methods”) before (ctrl) and after (MK801) drug injection. d–f Average gamma power (30–50 Hz) in frontal (d) and parietal cortex (e) and in hippocampus (f) elicited by electrical stimulation of the pontine reticular formation. Segments included in analysis in c–f are from simultaneous recordings. Calibration scale: a, b mV (same scale was used for all gamma traces), c–f 100 μV2
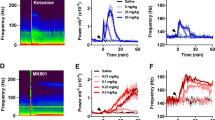
Next, we tested the acute effect of two NMDA-R antagonists in freely moving rats. Field potentials were simultaneously recorded at the two major theta dipoles, identified using the phase of theta waves relative to cortical EEG over the parietal cortex, i.e. in-phase and out-of-phase, respectively, for recordings at the CA1 pyramidal layer (CA1-theta) and at the hippocampal fissure at the termination of the perforant path input (DG-theta) (Buzsaki et al. 1986; Bragin et al. 1995; Kocsis et al. 1999; Montgomery et al. 2009). After injection of MK801 (0.2 mg/kg, s/c) or ketamine (10 mg/kg, s/c), gamma power drastically increased in both CA1 and DG and the effect lasted for ~1 and ~4 h, respectively (Fig. 2) (two-way ANOVA, pre vs. post-injection: F[1,49] = 41.65, P < 0.001, CA1 vs. DG: F[1,49] = 2.04, P = 0.16; interaction F[1,49] = 1.37, P = 0.25).

Acute effect of NMDA antagonists on hippocampal theta and gamma oscillations in freely moving rats. a, b Changes in theta power (a 5–10 Hz band and b theta peak) after injection of ketamine and MK801 (averaged over 30 min and 2 h, respectively) relative to 30 min control recorded during wake motor activity. c Peak theta frequency before and after injection. d–g Time course of average theta (5–10 Hz) and gamma (30–50 Hz) power in the CA1 (n = 9) and DG (n = 4) areas of the hippocampus before and after injection of MK801 (0.2 mg/kg, s/c; n = 9 and 4) and ketamine (10 mg/kg, s/c; n = 6) in freely moving rats. Dots represent theta and gamma power averaged over 5 min segments, normalized using 1 h averages in the beginning of the recording session. Note different time scales for MK801 and ketamine. h Theta and gamma averages in parietal cortex (n = 7). i Traces of theta and gamma power in the CA1 region in individual experiments (n = 6). Note large variation of theta before and 2 h after ketamine injection
In contrast, changes in theta showed regional specificity (time vs. area interaction: F[1,49] = 10.41, P = 0.002); theta power within the 5–10 Hz frequency range decreased in CA1 by 24 ± 7% after MK801 and 20 ± 6% after ketamine, but remained high in the DG (Fig. 2a). Changes in theta power in the CA1 followed the same time course as gamma activation, whereas theta in DG decreases after 30–60 min (Fig. 2d–g). Note that theta rhythm strongly depends on behavior and thus periods of high and low theta are alternating in the EEG, resulting in relatively high variation of group averages in control recordings (i.e. before and long after injection, Fig. 2d–g). After NMDA-R blockade, the rats were awake, and showed high motor activity which included constant locomotion, i.e. moving around in the recording box, circling, rearing, and stereotypic head movements (Manahan-Vaughan et al. 2008), accompanied with high neck muscle EMG. Theta was present during this motor activity at increased frequency (from 7.1 ± 0.1 to 8.4 ± 0.3 Hz after MK801 and from 6.9 ± 0.3 to 7.8 ± 0.3 Hz after ketamine) (Fig. 2c), at which the peak spectral power did not change (Fig. 2b), even though average 5–10 Hz power decreased in CA1 after drug injection in all rats, as shown in individual traces in Fig. 2i. Statistical comparison of post-injection theta power with theta during pre-injection wake periods confirmed theta decrease in CA1 (one-way ANOVA, F[1,29] = 10.42, P = 0.003) and indicated that DG theta power was at the same level as during active waking before injection (F[1,19] = 2.87, P = 0.11).
The changes in the parietal cortex, both in the theta and gamma ranges, were similar to those in the CA1 region of the hippocampus (Fig. 2h) (paired t test, theta: t[6] = 4.98, P = 0.002; gamma: t[6] = −4.35, P = 0.005). Gamma activity in the frontal cortex also increased (ketamine: t[7] = −8.07108, P < 0.001; MK801: t[8] = −5.21, P < 0.001), in agreement with previous studies (Pinault 2008; Hakami et al. 2009; Lazarewicz et al. 2010; Kocsis 2011).
The third experiment compared the acute effects of ketamine using different routes of systemic administration, i.e. intraperitoneal (i/p) and subcutaneous (s/c) injections. This step was necessary to address the apparent discrepancy between the dose of ketamine commonly used in the chronic schizophrenia model (30 mg/kg repeated injections i/p) (Becker et al. 2003) and the dose which induced “aberrant” gamma oscillations in acute experiments (10 mg/kg s/c) (Pinault 2008; Hakami et al. 2009). Thus, we injected ketamine i/p in different doses (10, 20, 40, 60 mg/kg) in seven rats and compared the changes in gamma power with those induced by 10 mg/kg s/c injection. Ketamine increased gamma power in either route of administration (Fig. 3). In frontal cortex, the effect of i/p injection was dose-dependent (ANOVA: F[3,13] = 7.622, P = 0.006) and, compared with 10 mg/kg s/c injection, the equivalent i/p dose was between 20 and 40 mg/kg (Fig. 3c) (P values: 0.001, 0.012, 0.52, and 0.014, respectively, for pair-wise comparisons of s/c 10 mg/kg vs. i/p 10, 20, 40, 60 mg/kg). Increased gamma oscillations lasted longer after i/p (2–3 h depending on dose, Fig. 3a) than after s/c injections (60–120 min). The changes were identical in the CA1 and similar in the DG with a somewhat less pronounced dose-dependent relationship at high doses of ketamine (Fig. 3d).

Comparison of the acute effect of i/p and s/c ketamine injection in freely moving rats. a Temporal dynamics of changes in gamma activity after i/p administration of ketamine in different doses (10, 20, 40, 60 mg/kg) and comparison with s/c injection. (“chronic” shows the acute effect of ketamine in chronic experiments). Average spectral power in the 30–50 Hz frequency range was calculated in 5 min windows. b Effect of i/p and s/c ketamine on spectral power in different frequency bands. Power averaged over the first 45 min after injection is expressed relative to 45 min control segment before injection. c, d Dose–response effect of i/p ketamine on gamma power in frontal cortex (c) and hippocampus (d) and comparison with the effect of s/c injection (gamma calculated as in b)
Hippocampal oscillations in the chronic ketamine model of schizophrenia
In chronic experiments, EEG recordings started 2 weeks before the first ketamine injection and lasted for 2–4 weeks after the treatment was completed. The acute effect of ketamine in these experiments was recorded on the first day of treatment in a subset of animals (n = 5) and was found within the range of the acute study (Fig. 3a, “chronic”). After the chronic treatment, theta oscillation showed a steady decline in the hippocampus (Fig. 4a, b). Two-way ANOVA revealed significant interaction (F[5,77] = 7.94, P = 0.049) between treatment (saline vs. ketamine) and time (three recordings before and three after treatment on days 2–3, 5–7 and 14–15). After 2 weeks, average theta power dropped to 45 ± 7% whereas after saline theta was still at 91 ± 6% of the control level. The changes were significant in the ketamine group (one-way ANOVA, F[5,53] = 9.20, P < 0.001; with significant differences between pre and post-treatment time points according to posthoc Bonferroni comparison) but not in the saline group (F[5,23] = 2.18, P = 0.10).

Effect of chronic treatment with subanesthetic doses of ketamine on hippocampal oscillations. a Group averages of hippocampal theta power in a total of six recording sessions during the week before (Pre) and during a 2-week period after a 5-day course of daily injection of ketamine or saline (wk0: day 2–3, wk1: day 5–7, wk2: day 14–15 after the end of treatment). Power was normalized and expressed as percent of the average of the three pre-treatment recordings. b Changes in hippocampal theta power in individual experiments. c Changes in hippocampal theta power during theta states associated with waking exploration (MOV) and REM sleep (REM). d Percent change in gamma (30–50 Hz) power in hippocampal EEG
Since the acute effect of ketamine on theta power varied between different layers of the hippocampus, we tested whether the location of the recording electrode modified the results of chronic treatment, as well. There was no significant treatment versus area (CA1 and DG) interaction (F[2,77] = 0.01, P = 0.92) in two-way ANOVA and the reduction in theta power was significant for recordings from either of these layers after ketamine (one-way ANOVA, P < 0.001) but not after saline injections. Similar relationships were also found for theta power in the parietal cortex (ketamine: F[5,53] = 6.8, P < 0.001; saline: F[5,23] = 1.92, P = 0.14).
Theta rhythm in rats has strong association with behavior. Synchronization of EEG power in the theta band occurs during waking motor activity (exploration) and REM sleep and there are important differences in the mechanisms of theta generation in these theta states (Bland 1986; Vertes and Kocsis 1997; Buzsaki 2002). Thus, we next investigated how theta power changes in these theta states after chronic treatment with ketamine. All EEG segments dominated by theta synchrony (theta vs. delta ratio above 4) (Csicsvari et al. 1999) were separated from the recordings, grouped as waking versus REM sleep (see “Methods”), and the power spectra of these segments were averaged over the daily recordings (Fig. 4c). Theta in REM sleep and waking was equally affected by ketamine treatment; after 2 weeks, theta power was less than 50% of the pre-treatment average in both behaviors. Two-way ANOVA revealed significant differences in theta over time (F[5,103] = 9.94, P < 0.001) but no significant effect of behavior (REM vs. wake, F[1,103] = 0.11, P = 0.74) and no significant interaction between time and behavior (F[5,103] = 0.26, P = 0.93).
Gamma activity also significantly decreased in the hippocampus (Fig. 4d). 2 weeks after ketamine treatment, gamma power was reduced by 38 ± 8%, as compared with 14 ± 11% in saline control (two-way ANOVA: treatment vs. time interaction: F[5,82] = 15.18, P = 0.004; effect of time: F[5,54] = 8.9, P < 0.001 for ketamine, F[5,27] = 1.38, P = 0.27 for saline). Gamma power in the frontal cortex EEG after 2 weeks was 82 ± 10% of pre-treatment control, but the pre- versus post-injection difference was not significant (F[5,54] = 1.8, P = 0.12) and was also not significantly different from saline control (two-way ANOVA: treatment vs. time interaction: F[5,82] = 0.59, P = 0.71).
Interindividual variability of the effect of chronic ketamine treatment on hippocampal oscillations
Ketamine treatment did not affect each rat equally (Fig. 4b). Figure 5 shows two experiments which represent characteristic patterns of the changes in hippocampal oscillations. In the first rat, there was a strong decrease in theta and gamma powers, whereas the other showed a relatively mild effect on theta and no change in gamma rhythm. Furthermore, in the first rat (Fig. 5a–c), the changes were already present by the end of the first week and did not change considerably afterward. In the second rat (Fig. 5d, e), theta declined progressively over a 2-week period. Similar patterns of decrease in theta power were found when the analysis only included EEG segments, in which theta was dominant (Fig. 5b, e), indicating that the changes in theta were due to the effect on the theta generator rather than to changes in behavior, i.e. decrease in theta could not be explained by shortening of theta episodes or decrease in their number. On the group level, inter-individual variability was tested using Levene’s test of equal variances at different time points. Group variance of hippocampal theta and gamma powers significantly increased after treatment compared with pre-injection measurements for both theta (F[1,21] = 12.1, P = 0.002) and gamma (F[1,21] = 14.9, P < 0.001) rhythms.

Patterns of the change in autospectra of hippocampal EEG after chronic treatment with ketamine in two rats (a–c, d–f). a, d Relative power in the 0–15 Hz frequency range before (pre) and after (post) ketamine treatment calculated over 5 h of continuous recording. b, e Average autospectra of all theta episodes of hippocampal EEG during each 5-h recording session. Note drop in theta power after ketamine treatment in the first rat (b) with no further change during the next 4 weeks and progressive decline in theta rhythm in the second rat (b) from day 5 (d5) to day 13 (d13) after the last day of treatment and. c, f Change in spectral power in the 1–50 Hz range. Note 40–50% decrease in gamma in the first experiment (c) and minor change in the second (f)
Next, we tested whether inter-individual differences in the extent of reduction in theta and gamma powers correlated with structural alterations in the network of PV+ interneurons in the hippocampus, in a representative subset of nine rats (3 saline and 6 ketamine). The changes in the electrophysiological parameters in this group were similar to those of the entire population of rats treated with ketamine and saline in this study. Theta and gamma power, 2 weeks after ketamine injections, were 58 ± 12% and 70 + 13%, respectively, of the pre-injection level (Fig. 7a) and showed large variation (range: 16–104% and 20–105% for theta and gamma, respectively) which overlapped with the parameters of the saline-treated subjects (93 ± 9%, range: 76–107% for theta and 97 ± 7%, range: 84–105% for gamma) (compare Figs. 4b and 7b).
The number of detectable PV+ cells in the CA1 region were significantly lower in the group of ketamine treated rats than in the saline control in both CA1 and CA3 (two sample t test, unequal variances, t[7] = 2.86, P = 0.025 for CA1 and t[7] = 3.87, P = 0.035 for CA3) (Table 1; Fig. 6), confirming previous reports (Keilhoff et al. 2004). In individual experiments, PV+ cell number was low in five of six ketamine treated rats, compared with control (Fig. 7c). One rat showed high PV+ cell number and moderate changes in theta (67% of pre-injection level) and gamma (88% of pre-injection level) oscillations, even though the effectiveness of the acute injection had been verified by acute gamma increase during treatment.

Differences in hippocampal parvalbumin positive interneurons in control (saline) and ketamine treated rats. Upper panels (Case A55) show a representative hippocampal section with darkly stained cell bodies and neuropil of a saline treated control rat, lower panels show an example of ketamine treated rat (Case A56). A higher magnification photo is shown on the right. Note fewer PV positive cell bodies in CA1 of A55 compared with A56

Comparison of loss of PV+ cells in the hippocampus and decrease in theta and gamma power. a Group averages of hippocampal CA1 theta and gamma power before (Pre) and after chronic ketamine treatment in six rats. b Changes in hippocampal CA1 theta power in individual experiments treated with saline (n = 3) or ketamine (n = 6). c Number of detectable PV+ cells in the CA1 area of the hippocampus of the same group of rats. d Scatter plot of theta (top) and gamma power (bottom) 2 weeks after ketamine treatment, expressed as percent of pre-injection average, versus PV+ cell number, in five rats which showed loss of PV+ cells
When analyzing interindividual differences, however, in the five rats which showed decreased PV+ cell numbers, we found that larger structural deficits did not necessarily lead to larger functional loss. In fact, surprisingly, the two parameters changed in opposite directions in the CA1, i.e. relatively more severe changes in PV cell number accompanied less severe oscillation deficits. Figure 7d shows the distribution of the PV+ cell number in CA1 versus the percent of theta and gamma power still present in individual rats in CA1 field potential recordings after ketamine treatment. The electrophysiological parameters showed negative correlation with PV expression in the CA1 region (R 2 = 0.53 (P = 0.075) for theta and R 2 = 0.78, (P = 0.006) for gamma; Spearman rank correlation, t[3] = −3.58, P = 0.037, for both) indicating that even though chronic administration of ketamine lead to parallel decreases in PV+ cell number and theta/gamma oscillations, within the ketamine treated group, a further PV decrease correlated with strengthening of these oscillations (Fig. 7d).
Discussion
This study compared the changes in hippocampal theta and gamma oscillations in the chronic ketamine model of schizophrenia with the effects of single injections of NMDA-R antagonists. We found that injection of MK801 or ketamine in freely moving rats increased gamma power in both CA1 and DG at short latencies, while theta alteration showed regional specificity. Theta peak shifted to higher frequencies whereas average 5–10 Hz theta power decreased in CA1 but remained high in the DG. Field potential oscillations in the parietal cortex also showed theta decrease and gamma increase, similar to that of the CA1 region of the hippocampus. The effects lasted ~1 and 4 h for ketamine and MK801, respectively. During this time, the rats were awake and exhibited stereotypic behavior accompanied by high neck muscle activity. A similar pattern of opposite changes in gamma versus theta oscillations in hippocampal CA1 and parietal cortex was also observed under urethane anesthesia where theta and gamma can be triggered by direct activation of the ascending drive from the reticular formation in a controlled manner, independent of behavior. In contrast, chronic administration of ketamine caused a steady decline in theta and gamma oscillations, over the 2–4 weeks after treatment, in both CA1 and DG. A further important difference between the two models was that the effects of acute injection were more robust than the changes after chronic treatment. Histological examination confirmed a decrease in the number of detectable PV+ interneurons in the chronic ketamine model (Keilhoff et al. 2004) but also revealed, unexpectedly, that within the ketamine treated group, a further PV decrease correlated with strengthening of these oscillations.
NMDA-R hypofunction has been linked to GABA pathology, i.e. to loss of GAD and PV expression (Gonzalez-Burgos and Lewis 2008; Lisman et al. 2008), which constitutes the most consistent postmortem histological finding in schizophrenia (Todtenkopf and Benes 1998; Reynolds et al. 2004; Lewis et al. 2005). PV+ interneurons are involved in the generation of oscillations suggesting a causal relationship between this histological marker and altered oscillations documented in human schizophrenia (Spencer et al. 2009; Uhlhaas and Singer 2010) and in a variety of animal models (Lodge et al. 2009; Kocsis 2011; Lanre-Amos and Kocsis 2010; Lazarewicz et al. 2010; Vohs et al. 2010). The decrease in theta and gamma oscillations in the group of chronic ketamine treated rats in the present study is in agreement with this proposition, as decrease in GAD67+ and PV+ cell number has been demonstrated in this model earlier (Keilhoff et al. 2004) and PV decrease was detected in our experiment.
Furthermore, we found relatively large variations in theta and gamma impairment in the chronic model as compared with acute injection, and the number of detectable PV+ cells also varied within the group of chronic treated rats. This allowed a more detailed analysis of the relationship between the two parameters, i.e. testing whether larger decreases in PV+ cell number would result in larger decrease in the power of oscillations. This hypothesis was, however, not supported by the results of the experiment. First, after ketamine treatment there was a single large drop [26 and 44% in CA1 and CA3, respectively cf. 39 and 34% in Keilhoff et al. (2004)] in the PV+ cell number in five of six rats (see in Fig. 7c) which did not match the relatively even distribution of theta and gamma power between no change in some rats and severe impairment in rats at the other end of the spectrum (see in Fig. 4b). Second, there was no indication of a parallel decline in PV+ cell number and oscillation power. In fact, we found the opposite, i.e. the more severe the PV+ loss the less severe the theta and gamma impairment. It should be noted, however, that the number of brains included in histology was relatively low and unfortunately, no data in the literature is available for comparison, as previous reports only included group statistics. Thus, understanding the relationship between the two parameters in this model will require further experiments in a larger number of animals. These will also have to include different time points as PV+ cell loss was still absent 72 h after the treatment, according to a recent study (Benneyworth et al. 2011), even though theta impairment started to manifest at that early stage in the present study (wk0 in Figs. 4, 7b). Assessment of both interindividual differences and time course may be critical for understanding subchronic and chronic models in which, just as in the human disease, it is difficult to separate alterations directly induced by the insult from those due to various compensatory processes. This may be especially true for chronic models, either genetic or generated by interventions in the early stages of life. Both PV+ cell loss and cognitive deficits are commonly present in these models (Moore et al. 2006; Harte et al. 2007; Tseng et al. 2008; Wen et al. 2010), and oscillations may also be impaired.
There are several mechanisms that can compensate for GABA deficit, especially after chronic interventions. Regulation of PV expression was proposed (Lewis et al. 2005) as one of these mechanisms acting in schizophrenia. Whereas, decrease in GABA transmission due to downregulation of GAD67 or loss of interneurons leads to attenuated oscillations, a further decrease in PV expression in the functioning synapses may facilitate GABA release during fast repetitive firing (Gonzalez-Burgos and Lewis 2008) and may restore some of the lost oscillations. Indeed, increased GABA release and enhanced gamma oscillations have been shown in PV-knockout mice (Vreugdenhil et al. 2003). Enhanced hippocampal oscillations were also observed in a subchronic model of schizophrenia (Lanre-Amos and Kocsis 2010) with documented decrease in detectable PV+ cells and terminals (Berretta et al. 2004). The relationship between the PV cell number and oscillatory power found in this study would also allow the interpretation that after the initial damage of the GABA network, additional PV downregulation may be compensatory.
Changes in GAD67 and PV expressions are far too slow (Kinney et al. 2006), however, to play a role in the acute reaction. The short latency of the effect of gamma activation and parallel theta suppression indicates that NMDA antagonists lead to rapid alteration in the activity of neuronal networks, which interfere with oscillations. Several classes of interneurons involved in rhythmogenesis are sensitive to NMDA-R antagonists, including PV+ basket and axo-axonic cells (Buhl et al. 1994, 1996; Grunze et al. 1996). The expression of NMDA-Rs by PV+ cells, however, shows a large variability (Nyiri et al. 2003) indicating that only a subset of these interneurons is affected. Disfacilitation of this population may increase the activity of pyramidal cells and thus enhance synchronous phasic activity in a reciprocally connected pyramidal cell-interneuron network (Middleton et al. 2008; Carlen et al. 2011).
Changes in theta rhythm may be due to the effect of NMDA antagonists on a different class of interneurons located in the stratum oriens and projecting to the lacunosum molecular layer (O-LM). These cells also express PV+ and are very sensitive to NMDA blockade (Hajos et al. 2002). They are not synchronized with gamma oscillations (Klausberger et al. 2003) but play a critical role in generating theta rhythm (Gillies et al. 2002; Klausberger et al. 2003). Theta–gamma coordination is important for cognitive processes (Lakatos et al. 2008; Lisman and Buzsaki 2008) and O-LM interneurons were shown to critically contribute to the formation of gamma-coherent cell assemblies at long distances by entrainment at theta frequency (Tort et al. 2007). Changes in hippocampal oscillations may also develop due to mechanisms potentially related to glutamate receptors outside of the hippocampus. Theta power and frequency is regulated by a number of subcortical structures, including most importantly the pontine reticular formation, median raphe, supramammillary nucleus, and medial septum (Vertes and Kocsis 1997). The involvement of medial septum and supramammillary nucleus in NMDA-R antagonist induced gamma and impairment of prepulse inhibition has been demonstrated (Ma and Leung 2007). In the brainstem, however, injection of MK-801 or AP-7 in the pontine reticular formation, normally promoting hippocampal theta, had no effect whereas NMDA-R blockade of the median raphe nucleus, normally inhibiting theta, was shown to produce theta rhythm in the hippocampus at short latencies and long duration, in urethane anesthetized rats (Kinney et al. 1994).
The opposite changes in gamma power notwithstanding, schizophrenia-relevant cognitive deficits were shown after both subchronic (Becker et al. 2003; Stefani and Moghaddam 2005; Rujescu et al. 2006) and acute (Mansbach and Geyer 1989; Manahan-Vaughan et al. 2008; Kocsis 2011) NMDA-R blockade supporting an “aberrant” nature of gamma activity induced by NMDA-R antagonists (Pinault 2008; Hakami et al. 2009). A critical attribute of the aberrant gamma oscillation may be the loss of theta rhythm and the consequent reduction of gamma coordination. There may be other alternative mechanisms, however, of how changes in oscillation power results in impaired neuronal synchrony which thus require further investigation using direct recordings of the activity of single neurons and neuronal ensembles and mathematical modeling. Furthermore, although proper organization and function of GABAergic interneuron networks is essential for many cognitive processes, the present data should not be interpreted as indicating that all cognitive deficits due to NMDA-R blockade and chronic hypofunction are the direct consequences of altered oscillations. For example, it has been shown recently, that mice lacking NMDA-Rs on PV+ in interneurons showed cognitive deficits in some tasks (e.g. associative learning) but not in others (e.g. prepulse inhibition on startle and water maze) (Carlen et al. 2011).
Validity for human schizophrenia
The key functional observations supporting the NMDA-R hypofunction model of schizophrenia arise from the acutely generated state after NMDA-R antagonist injection, which resembles most symptoms of the chronic disease. These include not only psychotic (Javitt and Zukin 1990) but also cognitive and negative symptoms (Krystal et al. 1994; Adler et al. 1999). Neuropathologic and genetic evidence of NMDA-R involvement in schizophrenia, however, is mostly indirect, with few notable exceptions such as the reduced NR2A receptors in interneurons of schizophrenics (Woo et al. 2004) or the impairment of the NMDA-R-mediated mismatch negativity (Javitt et al. 1995, 1996). NMDA-R mRNA and receptor binding studies only showed modest changes (Meador-Woodruff and Healy 2000), in contrast to ample evidence implicating NMDA-R in the pathomechanism of the well-described and consistent pattern of GABA pathology (Lewis et al. 2005; Lisman et al. 2008). Similarly, susceptibility genes for schizophrenia identified by genetic linkage studies encode proteins which, among their other functions, modulate the expression or the subcellular effect of NMDA-R activation rather than evoke gross abnormalities in the receptor (Moghaddam 2003).
Interpretation of the validity of ketamine induced acute gamma increase in humans (Plourde et al. 1997; Hong et al. 2010) is also not straightforward, as in schizophrenia patients most studies report decreased oscillations (Spencer et al. 2009; Uhlhaas and Singer 2010). Our study showed both enhancement and suppression of gamma after ketamine administration, depending on whether it was tested in the acute state or weeks after chronic treatment. It is tempting to interpret these findings as indicating that in schizophrenia gamma increase and decrease may both be a consequence of NMDA-R hypofunction and that these tendencies may coexist in different stages of the disease. Indeed, although EEG recordings during psychotic episodes are extremely rare, anecdotal data showed elevated gamma activity in this state (Baldeweg et al. 1998). Enhanced background gamma activity may also be present in the relatively stable “chronic” state but masked by impairment of task related oscillations, which is the standard focus of such studies. This has been shown recently in rats where induced and evoked gamma, associated with auditory stimulation, decreased while background gamma activity increased after acute ketamine injection (Lazarewicz et al. 2010) and in chronic models (Vohs et al. 2010; Carlen et al. 2011).
In humans, coexistence of mechanisms increasing and decreasing gamma oscillations were detected by Spencer et al. (2009) using acoustic stimuli applied to a heterogeneous patient population in which the composition of schizophrenia symptoms varied between subjects. The auditory steady state response to 40 Hz stimuli was tested in the patient group and correlated with the severity of positive symptoms. This latter was scored using a hallucination questionnaire and reflected the patients’ experience prior to the study and was thus unrelated to the recordings. They found an overall gamma reduction in schizophrenics compared with healthy controls but the reduction within the patient group negatively correlated with the severity of hallucinations, i.e. more severe schizophrenia symptoms lead to less severe reduction of the propensity to ~40 Hz oscillations. This pattern of the changes in the capacity of oscillatory networks is similar to our findings on how impaired oscillations were related to PV+ cell loss, another possible marker of the severity of the schizophrenia-relevant structural damage.
Conclusion
Both acute and chronic NMDA-R blockade have been used in previous studies to validate the NMDA-R hypofunction model. These investigations found that the changes in interneurons (Keilhoff et al. 2004; Kinney et al. 2006) and in cognition (Mansbach and Geyer 1989; Becker et al. 2003; Stefani and Moghaddam 2005; Rujescu et al. 2006; Manahan-Vaughan et al. 2008) were similar in the two types of models and consistent with human data. Observations in EEG oscillations which may serve as a link between the pathology of GABA networks and cognition deficits presents a unique challenge, however, as ketamine induced acute increase has to be reconciled with chronic decrease of gamma activity in schizophrenics. The present study demonstrates that NMDA-R blockade can lead to both these conditions and thus validate the NMDA-R hypofunction model in this domain.
References
Adler CM, Malhotra AK, Elman I, Goldberg T, Egan M, Pickar D, Breier A (1999) Comparison of ketamine-induced thought disorder in healthy volunteers and thought disorder in schizophrenia. Am J Psychiatry 156:1646–1649
Baldeweg T, Spence S, Hirsch SR, Gruzelier J (1998) Gamma-band electroencephalographic oscillations in a patient with somatic hallucinations. Lancet 352:620–621
Becker A, Grecksch G (2004) Ketamine-induced changes in rat behaviour: a possible animal model of schizophrenia. Test of predictive validity. Prog Neuropsychopharmacol Biol Psychiatry 28:1267–1277
Becker A, Peters B, Schroeder H, Mann T, Huether G, Grecksch G (2003) Ketamine-induced changes in rat behaviour: a possible animal model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 27:687–700
Benes FM, Kwok EW, Vincent SL, Todtenkopf MS (1998) A reduction of nonpyramidal cells in sector CA2 of schizophrenics and manic depressives. Biol Psychiatry 44:88–97
Benneyworth MA, Roseman AS, Basu AC, Coyle JT (2011) Failure of NMDA receptor hypofunction to induce a pathological reduction in PV-positive GABAergic cell markers. Neurosci Lett 488:267–271
Berretta S, Lange N, Bhattacharyya S, Sebro R, Garces J, Benes FM (2004) Long-term effects of amygdala GABA receptor blockade on specific subpopulations of hippocampal interneurons. Hippocampus 14:876–894
Bland BH (1986) Physiology and pharmacology of hippocampal formation theta rhythms. Prog Neurobiol 26:1–54
Bragin A, Jando G, Nadasdy Z, Hetke J, Wise K, Buzsaki G (1995) Gamma (40–100 Hz) oscillation in the hippocampus of the behaving rat. J Neurosci 15:47–60
Braun I, Genius J, Grunze H, Bender A, Moller HJ, Rujescu D (2007) Alterations of hippocampal and prefrontal GABAergic interneurons in an animal model of psychosis induced by NMDA receptor antagonism. Schizophr Res 97:254–263
Buhl EH, Szilagyi T, Halasy K, Somogyi P (1996) Physiological properties of anatomically identified basket and bistratified cells in the CA1 area of the rat hippocampus in vitro. Hippocampus 6:294–305
Buhl EH, Han ZS, Lorinczi Z, Stezhka VV, Karnup SV, Somogyi P (1994) Physiological properties of anatomically identified axo-axonic cells in the rat hippocampus. J Neurophysiol 71:1289–1307
Buzsaki G (2002) Theta oscillations in the hippocampus. Neuron 33:325–340
Buzsaki G, Draguhn A (2004) Neuronal oscillations in cortical networks. Science 304:1926–1929
Buzsaki G, Czopf J, Kondakor ILK (1986) Laminar distribution of hippocampal rhythmic slow activity (RSA) in the behaving rat: current source density analysis. Brain Res 365:125–137
Buzsaki G, Horvath Z, Urioste R, Hetke J, Wise K (1992) High-frequency network oscillation in the hippocampus. Science 256:1025–1027
Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, Ruhlmann C, Jones SR, Deisseroth K, Sheng M, Moore CI, Tsai LH (2011) A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry (epub ahead of print)
Csicsvari J, Hirase H, Czurkó A, Mamiya A, Buzsaki G (1999) Oscillatory coupling of hippocampal pyramidal cells and interneurons in the behaving rat. J Neurosci 19:274–287
Gao XM, Sakai K, Roberts RC, Conley RR, Dean B, Tamminga CA (2000) Ionotropic glutamate receptors and expression of N-methyl-d-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am J Psychiatry 157:1141–1149
Gillies MJ, Traub RD, LeBleau FEN, Davies CH, Gloveli T, Buhl EH, Whittington MA (2002) A model of atropine-resistant theta oscillations in rat hippocampal area CA1. J Physiol (London) 543:779–793
Goff DC, Coyle JT (2001) The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 158:1367–1377
Gonzalez-Burgos G, Lewis DA (2008) GABA neurons and the mechanisms of network oscillations: implications for understanding cortical dysfunction in schizophrenia. Schizophr Bull 34:944–961
Grunze HC, Rainnie DG, Hasselmo ME, Barkai E, Hearn EF, McCarley RW, Greene RW (1996) NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci 16:2034–2043
Hajos M, Hoffman WE, Kocsis B (2008) Activation of cannabinoid-1 receptors disrupts sensory gating and neuronal oscillations: relevance to schizophrenia. Biol Psychiatry 63:1075–1083
Hajos N, Freund TF, Mody I (2002) Comparison of single NMDA receptor channels recorded on hippocampal principal cells and oriens/alveus interneurons projecting to stratum lacunosum-moleculare (O-LM cells). Acta Biol Hung 53:465–472
Hakami T, Jones NC, Tolmacheva EA, Gaudias J, Chaumont J, Salzberg M, O’Brien TJ, Pinault D (2009) NMDA receptor hypofunction leads to generalized and persistent aberrant gamma oscillations independent of hyperlocomotion and the state of consciousness. PLoS One 4:e6755
Harte MK, Powell SB, Swerdlow NR, Geyer MA, Reynolds GP (2007) Deficits in parvalbumin and calbindin immunoreactive cells in the hippocampus of isolation reared rats. J Neural Transm 114:893–898
Hong LE, Summerfelt A, Buchanan RW, O’Donnell P, Thaker GK, Weiler MA, Lahti AC (2010) Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology 35:632–640
Javitt DC, Zukin SR (1990) The role of excitatory amino acids in neuropsychiatric illness. J Neuropsychiatry Clin Neurosci 2:44–52
Javitt DC, Doneshka P, Grochowski S, Ritter W (1995) Impaired mismatch negativity generation reflects widespread dysfunction of working memory in schizophrenia. Arch Gen Psychiatry 52:550–558
Javitt DC, Steinschneider M, Schroeder CE, Arezzo JC (1996) Role of cortical N-methyl-d-aspartate receptors in auditory sensory memory and mismatch negativity generation: implications for schizophrenia. Proc Natl Acad Sci USA 93:11962–11967
Kantrowitz JT, Javitt DC (2010) N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull 83:108–121
Keilhoff G, Becker A, Grecksch G, Wolf G, Bernstein HG (2004) Repeated application of ketamine to rats induces changes in the hippocampal expression of parvalbumin, neuronal nitric oxide synthase and cFOS similar to those found in human schizophrenia. Neuroscience 126:591–598
Kilts CD (2002) The changing roles and targets for animal models of schizophrenia. Biol Psychiatry 50:845–855
Kinney GG, Kocsis B, Vertes RP (1994) Injections of excitatory amino acid antagonists into the median raphe nucleus produce hippocampal theta rhythm in the urethane-anesthetized rat. Brain Res 654:96–104
Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM (2006) A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci 26:1604–1615
Klausberger T, Magill PJ, Marton LF, Roberts DB, Cobden PM, Buzsaki G, Somogyi P (2003) Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature 421:844–848
Kocsis B (2011) Differential role of NR2A and NR2B subunits in NMDA receptor antagonist-induced aberrant cortical gamma oscillations. Biol Psychiatry (in press)
Kocsis B, Li S (2004) In vivo contribution of h-channels in the septal pacemaker to the generation of theta rhythm in the rat. Eur J Neurosci 20:2149–2158
Kocsis B, Bragin A, Buzsaki G (1999) Interdependence of multiple theta generators in the hippocampus: a partial coherence analysis. J Neurosci 19:6200–6212
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB Jr, Charney DS (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51:199–214
Lakatos P, Karmos G, Mehta AD, Ulbert I, Schroeder CE (2008) Entrainment of neuronal oscillations as a mechanism of attentional selection. Science 320:110–113
Lanre-Amos T, Kocsis B (2010) Hippocampal oscillations in the rodent model of schizophrenia induced by amygdala GABA receptor blockade. Front Psychiatry 1:132
Lazarewicz MT, Ehrlichman RS, Maxwell CR, Gandal MJ, Finkel LH, Siegel SJ (2010) Ketamine modulates theta and gamma oscillations. J Cogn Neurosci 22:1452–1464
Lewis DA, Hashimoto T, Volk DW (2005a) Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 6:312–324
Lisman J, Buzsaki G (2008) A neural coding scheme formed by the combined function of gamma and theta oscillations. Schizophr Bull 34:974–980
Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA (2008) Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci 31:234–242
Lodge DJ, Behrens MM, Grace AA (2009) A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci 29:2344–2354
Ma J, Leung LS (2007) The supramammillo-septal-hippocampal pathway mediates sensorimotor gating impairment and hyperlocomotion induced by MK-801 and ketamine in rats. Psychopharmacology (Berl) 191:961–974
Manahan-Vaughan D, von Haebler D, Winter C, Juckel G, Heinemann U (2008) A single application of MK801 causes symptoms of acute psychosis, deficits in spatial memory, and impairment of synaptic plasticity in rats. Hippocampus 18:125–134
Mansbach RS, Geyer MA (1989) Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology 2:299–308
Marcotte ER, Pearson DM, Srivastava LK (2001) Animal models of schizophrenia: a critical review. J Psychiatry Neurosci 26:395–410
Meador-Woodruff JH, Healy DJ (2000) Glutamate receptor expression in schizophrenic brain. Brain Res Brain Res Rev 31:288–294
Medoff DR, Holcomb HH, Lahti AC, Tamminga CA (2001) Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus 11:543–550
Middleton S, Jalics J, Kispersky T, Lebeau FE, Roopun AK, Kopell NJ, Whittington MA, Cunningham MO (2008) NMDA receptor-dependent switching between different gamma rhythm-generating microcircuits in entorhinal cortex. Proc Natl Acad Sci USA 105:18572–18577
Moghaddam B (2003) Bringing order to the glutamate chaos in schizophrenia. Neuron 40:881–884
Moghaddam B, Jackson ME (2003) Glutamatergic animal models of schizophrenia. Ann N Y Acad Sci 1003:131–137
Montgomery SM, Betancur MI, Buzsaki G (2009) Behavior-dependent coordination of multiple theta dipoles in the hippocampus. J Neurosci 29:1381–1394
Moore H, Jentsch JD, Ghajarnia ME, Geyer MA, Grace AA (2006) A neurobehavioral systems analysis of the effects of gestational exposure to methylazoxymethanol acetate (MAM) in the rat: implications for the neuropathology of schizophrenia. Biol Psychiatry 60:253–264
Morrow BA, Elsworth JD, Roth RH (2007) Repeated phencyclidine in monkeys results in loss of parvalbumin-containing axo-axonic projections in the prefrontal cortex. Psychopharmacology (Berl) 192:283–290
Nyiri G, Stephenson FA, Freund TF, Somogyi P (2003) Large variability in synaptic N-methyl-d-aspartate receptor density on interneurons and a comparison with pyramidal-cell spines in the rat hippocampus. Neuroscience 119:347–363
Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates. Elsevier Academic Press, Amsterdam
Pinault D (2008) N-methyl-d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol Psychiatry 63:730–735
Plourde G, Baribeau J, Bonhomme V (1997) Ketamine increases the amplitude of the 40-Hz auditory steady-state response in humans. Br J Anaesth 78:524–529
Reynolds GP, Abdul-Monim Z, Neill JC, Zhang ZJ (2004) Calcium binding protein markers of GABA deficits in schizophrenia—postmortem studies and animal models. Neurotox Res 6:57–61
Rujescu D, Bender A, Keck M, Hartmann AM, Ohl F, Raeder H, Giegling I, Genius J, McCarley RW, Moller HJ, Grunze H (2006) A pharmacological model for psychosis based on N-methyl-d-aspartate receptor hypofunction: molecular, cellular, functional and behavioral abnormalities. Biol Psychiatry 59:721–729
Spencer KM, Niznikiewicz MA, Nestor PG, Shenton ME, McCarley RW (2009) Left auditory cortex gamma synchronization and auditory hallucination symptoms in schizophrenia. BMC Neurosci 10:85
Stefani MR, Moghaddam B (2005) Transient N-methyl-d-aspartate receptor blockade in early development causes lasting cognitive deficits relevant to schizophrenia. Biol Psychiatry 57:433–436
Tamminga CA, Stan AD, Wagner AD (2010) The hippocampal formation in schizophrenia. Am J Psychiatry 167:1178–1193
Todtenkopf MS, Benes FM (1998) Distribution of glutamate decarboxylase65 immunoreactive puncta on pyramidal and nonpyramidal neurons in hippocampus of schizophrenic brain. Synapse 29:323–332
Tort AB, Rotstein HG, Dugladze T, Gloveli T, Kopell NJ (2007) On the formation of gamma-coherent cell assemblies by oriens lacunosum-moleculare interneurons in the hippocampus. Proc Natl Acad Sci USA 104:13490–13495
Tseng KY, Lewis BL, Hashimoto T, Sesack SR, Kloc M, Lewis DA, O’Donnell P (2008) A neonatal ventral hippocampal lesion causes functional deficits in adult prefrontal cortical interneurons. J Neurosci 28:12691–12699
Uhlhaas PJ, Singer W (2010) Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci 11:100–113
Vertes RP, Kocsis B (1997) Brainstem-diencephalo-septohippocampal systems controlling the theta rhythm of the hippocampus. Neuroscience 81:893–926
Vohs JL, Chambers RA, Krishnan GP, O’Donnell BF, Berg S, Morzorati SL (2010) GABAergic modulation of the 40 Hz auditory steady-state response in a rat model of schizophrenia. Int J Neuropsychopharmacol 13:487–497
Vreugdenhil M, Jefferys JG, Celio MR, Schwaller B (2003) Parvalbumin-deficiency facilitates repetitive IPSCs and gamma oscillations in the hippocampus. J Neurophysiol 89:1414–1422
Wang XJ (2010) Neurophysiological and computational principles of cortical rhythms in cognition. Physiol Rev 90:1195–1268
Wen L, Lu YS, Zhu XH, Li XM, Woo RS, Chen YJ, Yin DM, Lai C, Terry AV Jr, Vazdarjanova A, Xiong WC, Mei L (2010) Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc Natl Acad Sci USA 107:1211–1216
Woo TU, Walsh JP, Benes FM (2004) Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-d-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry 61:649–657
Xi D, Zhang W, Wang HX, Stradtman GG, Gao WJ (2009) Dizocilpine (MK-801) induces distinct changes of N-methyl-d-aspartic acid receptor subunits in parvalbumin-containing interneurons in young adult rat prefrontal cortex. Int J Neuropsychopharmacol 12:1395–1408
Young JW, Powell SB, Risbrough V, Marston HM, Geyer MA (2009) Using the MATRICS to guide development of a preclinical cognitive test battery for research in schizophrenia. Pharmacol Ther 122:150–202
Zhang ZJ, Reynolds GP (2002) A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr Res 55:1–10
Acknowledgments
This study was supported by National Institute of Health Grants MH0083199 and by an investigator-initiated unrestricted grant by Sepracor, Inc.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kittelberger, K., Hur, E.E., Sazegar, S. et al. Comparison of the effects of acute and chronic administration of ketamine on hippocampal oscillations: relevance for the NMDA receptor hypofunction model of schizophrenia. Brain Struct Funct 217, 395–409 (2012). https://doi.org/10.1007/s00429-011-0351-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00429-011-0351-8