
Abstract
MRI was employed to follow the neurodegenerative foci and the localization of inflammatory cells by magnetically labeled CD4+ or CD8+ lymphocytes in the ischemia/reperfusion long-lived rats (9 and 13 months after 10 min of cardiac arrest). MRI of ischemic rats showed: (1) blood–brain barrier (BBB) leakage in the area of the dorsal hippocampus and brainstem-hindbrain level in basal cerebellum, (2) unlike anti-CD8 magnetic antibodies anti-CD4 ultra small paramagnetic iron oxide particles (USPIO) antibodies revealed hypointense areas in the brainstem-interbrain region and caudoputamen not found in animals that were not injected with USPIO antibodies, and (3) dilation in the retrosplenial area. Immunocytochemistry revealed microglial activation in the hippocampus and striatum, with indications of activation in thalamic lateral dorsal nuclei and the subventricular zone. In the CA1 and CA3 regions, it was noted that OX42- and ED1-positive granules appear in neuronal somata. Immunostaining of lymphocytes with TCR confirmed the T-cell presence in ischemic brain parenchyma of the hippocampus and striatum. The above observations thus point to a persistent dysfunction of BBB that in long-term may still lead to infiltration of T cells that are predominantly of helper (CD4+) type. Such inflammatory processes are backed by microglial activity even up to 1 year after ischemia/reperfusion. Moreover, in these animals an augmented expression of neurogenesis markers and neuroblast migration was also revealed in the subventricular zone. Thus, a balance of degenerative processes and inflammatory surveillance with neurogenesis could determine the long-term outcome of global ischemia survival or the previously proposed formation of amyloid plaques and Alzheimer’s-type dementia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ischemic brain injury is, in part, an inflammatory disease. Postischemic acute inflammatory cascade has been observed in the experimental and clinical conditions and is associated with the upregulation of intracellular adhesion molecules (Selakovic et al. 2009), which are involved in vascular adhesion, transendothelial migration of white blood cells through the open blood–brain barrier (BBB), and the upregulation and release of cytokines (Touzani et al. 2002; Saleh et al. 2004a; Pluta et al. 2009). These reactions participate in complex interlocking of signaling pathways, which ultimately upregulate or downregulate inflammatory processes of the host response to injury. Although neuroinflammation is a frequent culprit of brain ischemia (Stoll et al. 1998; Touzani et al. 2002; Saleh et al. 2004a; Yilmaz et al. 2006; Pluta et al. 2009) its complex role following brain ischemia remains unclear.
Primary acute brain ischemic injury is associated with chronic secondary molecular and cellular events that are initiated following ischemia and may last for months up to years (Kokaia et al. 2006; Thored et al. 2006; Pluta et al. 2009). These secondary processes interact in a complex network processing signal bias toward apoptosis or toward cell survival pathways in cellular targets (Sugawara et al. 2004). While neuroinflammation was initially thought to arise as secondary to ischemic degeneration, the recent studies reveal that inflammation may stimulate amyloid precursor protein metabolism by the upregulation of β-secretase (Sastre et al. 2003) thus establishing a particular vicious cycle. Prognosis of recovery after brain ischemia is worsened by the secondary brain injury that finally contributes to poor outcome with neurodegeneration and dementia (Ziv et al. 2006; Yang and Simpkins 2007; Pluta et al. 2009; Kiryk et al. 2011).
Interest in the role of inflammatory cells in neurological diseases has been constantly increasing, and it has become apparent that the central nervous system (CNS) is capable of mounting a sustained and robust inflammatory response in conditions such as brain ischemia (Touzani et al. 2002; Saleh et al. 2004a; Pluta et al. 2009) or pathologies such as Alzheimer’s disease (Arshavsky 2010). The question whether neuroinflammation is destructive and/or beneficial for a long time following brain ischemia remains unanswered. The above inflammatory reaction has previously been studied in acute models of brain ischemia (Touzani et al. 2002). Here, we study the inflammatory reaction in chronic ischemic brain injury. The present study was designed to follow cellular markers of inflammation in the brain in response to ischemia with a long-term survival (Pluta et al. 1991). To this aim the chronic effect of global cerebral ischemia on the inflammatory cellular interactions in the rat brain was followed 1 year after the insult by in vivo magnetic resonance imaging (MRI) with magnetically labeled anti-CD4 and anti-CD8 antibodies complemented by immunocytochemistry (macrophages/microglia with ED1 and OX42, and lymphocytes with TCR). In addition, since it was proposed that neurogenesis could be an important survival factor in global ischemia (Darsalia et al. 2005), we have examined if in these animals the ischemic insult has also induced persistent neurogenesis markers observable even 1 year after the insult.
Materials and methods
Animal model
Two-month-old female rats (Wistar, 160–180 g) were submitted to 10 min duration of global cerebral ischemia induced by cardiac arrest (Pluta et al. 1991). Groups of four animals per cage (Erath, FRG), were housed in an air-conditioned room, at a temperature of 23 ± 2°C, with 55 ± 10% humidity, and with lights on 12 h/day (7:00–19:00 hours). The animals were given commercial food and tap water ad libitum. All experimental procedures were performed during the light phase, between 9:00 and 15:00 hours under the identical conditions. Animals used for these procedures were treated in strict accordance with the NIH Guide for Care and Use of Laboratory Animals (1985) and European Community Council Directive (86/609/EEC), as well as with the approval of the local Ethical Committee. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Experimental procedure—induction of global cerebral ischemia
Global ischemia occurs when cerebral blood flow is reduced throughout all of the brain. Our animal model of global cerebral ischemia clinically represents reversible cardiac arrest. Global cerebral ischemia was performed by cardiac arrest of 10 min duration (Pluta et al. 1991). The animals were allowed to survive 9 and 13 months after reperfusion. Sham-operated rats were exposed to the same procedures as ischemic animals but without induced cardiac arrest and thus served as controls.
MR imaging
MRI was performed on 9 and 13 months postischemic rats (seven ischemic and seven control animals) using the clinical 1.5 T Avanto MR Imager (Siemens) and small surface RF coils. Rats were anaesthetized with tiletamine and zolazepam (“Zoletil”; 0.5 mg/kg) and placed in the prone position for MRI. Standard T1-weighted, T1W (with repetition time, TR relation to time to echo, TR/TE = 30/5 ms; field of view, FoV = 132 × 132) and Turbo Spin Echo, TSE T2W (5,000/83 ms; FoV = 131 × 131) sequences were used. Coronal images were obtained with slice thickness of 1.5 mm (voxel dimensions 1.5 × 0.2 × 0.3 mm). Postcontrast T1W images were obtained following i.v. injection of Gd-DTPA (“Magnevist”, 0.2 mmol/kg, Schering) to test BBB changes in vivo. The number of averages was adjusted to the acceptable S/N ratio, allowing a total scanning time less than 40 min. Following MRI scans, rats were warmed and monitored until they fully recovered.
To assess the infiltration of inflammatory cells in the brain tissue antibodies against CD4+ and CD8+ cells magnetically labeled with ultra small paramagnetic iron oxide particles—USPIOs (MACS®, Miltenyi Biotec) were i.v. injected (0.2 ml USPIO with 0.3 ml saline) into rats 20–24 h prior to imaging. 3D T2*W images were obtained using a gradient echo sequence (TR/TE = 100/25 and 100/35 ms; FoV = 132 × 132). The prolonging of the TE in the T2* sequence augments the signal artifact generated by USPIO, and thus serves as a reliable indicator of label’s presence in the tissue. USPIO was not injected to control animals since we relied on the previous work (Pirko et al. 2003) that demonstrated the absence of USPIO-like signals in naive animals. It is worth noting that we have kept the original definition of MACS® particles as USPIO according to Pirko et al. (2003) who first applied them (see in “Results”), although their dimension, 50 nm, is on the border of small paramagnetic iron oxide particles (SPIO). These nanoparticles are specific for being actually cross-linked iron oxide (CLIO) particles to IgG proteins—antibodies (Stoll and Bendszus 2009). This makes them more specific to the particular cell type as compared to ordinary (U)SPIO particles that get phagocytosed by reactive immune cells. The cells that take up these nanoparticles are macrophages/microglia, while T cells are left unlabeled. In the case of live monitoring of T cells, the only solution is offered by CLIO particles as employed in the present study.
Immunocytochemistry
Immunocytochemistry was performed 13 months after the ischemic insult on six ischemic and six control animals. After transcardial perfusion with 4% paraformaldehyde, the brains were postfixed in the same solution and cryoprotected in 30% sucrose. After freezing at −80°C, brains were cut on a cryostat in 30-μm-thick coronal slices. Cell antigens were labeled for activated microglia—anti-OX42 (1:100, Caltag laboratories), and macrophages/microglia—anti-ED1 (1:100, AbD Serotec). Visualization of primary labeling was done with Alexa Fluor 555-conjugated goat polyclonal anti-mouse antibodies (1:200, Molecular Probes). Neurons were labeled with fluroscent Nissl stain-NeuroTrace (1:100, Molecular Probes). This double staining was used for better morphological assessment of neurons versus activated microglia/macrophages. In addition, monoclonal mouse anti-rat TCR α/β biotin antibody (1:10, AbD Serotec) was used for the visualization of T lymphocytes and monoclonal rat anti-mouse CD31 (PECAM-1) antibody (1:500, BD Pharmingen) for the visualization of blood vessel endothelial cells. Secondary antibodies were FITC-streptavidin (1:50, Vector Laboratories) and Cy5-conjugated goat anti-rat antibodies (1:500, Jackson Immuno Research Laboratories), respectively. Further, monoclonal mouse anti-Ki67 antibody (1:10, Invitrogen) was used in order to assess the level of proliferation of neuroepithelial cells in the subventricular zone (SVZ), while the polyclonal rabbit anti-doublecortin (DCX) antibody was used for the determination of the neuronal progenitor cell population in the SVZ or the dentate subgranular zone of the hippocampus (1:500 or 1:250, respectively, Abcam). Secondary antibodies were Alexa Fluor 555-conjugated goat polyclonal anti-mouse and Alexa Fluor 555-conjugated goat polyclonal anti-rabbit antibodies, respectively (1:100, Molecular Probes).
Imaging was done using a confocal laser scanning microscope (LSM 510, Carl Zeiss GmbH) with an argon laser (488 nm) utilized for the excitation of Nissl staining and FITC-streptavidin, and helium–neon lasers (543 and 633 nm), for the excitation of Alexa Fluor 555 and Cy5, respectively. Objectives used were Plan-Neofluar 20×/0.5 and Plan-Apochromat 63/1.4 Oil DIC. Following acquisition, images were processed using the Zeiss LSM 510 Basic software package version 3.2.
In order to quantify fluorescence intensities in dual channel recordings, the ratio between the signal intensities in two confocal channels was performed by multiplying each signal pixel intensity by the number of pixels and adding these values for each pixel intensity for each channel. Then the ratio of total signal intensities for the two channels was calculated. This was performed with confocal images taken from brain slices with the objective magnification of 20×. Finally, these values were statistically compared with the Student’s unpaired t test using SigmaPlot 11.0 software package (Systat Software, Inc.).
Results
Magnetic resonance imaging was employed on the brains of anesthetized live ischemia/reperfusion long-lived (up to 13 months after reperfusion) rats in order to trace the infiltration of inflammatory cells, CD4+ or CD8+ T cells, with USPIO-tagged antibodies. This novel method was introduced by Pirko et al. (2003) in experimental allergic encephalomyelitis mice. Based on the T2* contrast, it was possible to visualize immune cell location in CNS. The MRI protocol relied on the fact that accumulated superparamagnetic nanoparticles—USPIOs induce susceptibility artifacts that are manifested as signal voids—hypointense zones in MR images. The area of the signal void is larger than the actual size of the tissue that accumulates USPIOs allowing that small amounts of these superparamagentic nanoparticles can be detected.
In order to target more close cellular markers of neuroinflammation, MRI-scanned animals and naive ones were killed for CNS tissue immunocytochemistry. Brain sections were stained for activated microglia, macrophages, and T cells, and analyzed by confocal laser scanning microscopy. In addition, the neuroblast activation and migration in the SVZ of long-term surviving rats as compared to sham controls were identified by staining for proliferation-associated markers Ki67 and DCX.
Foci of inflammation and the state of the BBB
MRI was employed to check for the infiltration of immune cells in the brain parenchyma in living animals 1 day after i.v. injection of antibodies against CD4 or CD8 lymphocyte receptors (see examples of obtained images in Fig. 1a, b). In four tested animals (13 months after insult), anti-CD4 magnetic antibodies specific for the helper T cells revealed hypointense areas in the brainstem-interbrain region/caudoputamen-striatum likely to originate from paramagnetic iron since no such signal was found in sham-operated animals not injected with these antibodies (n = 7). In addition, prolonging the echo time in the gradient echo T2* sequence (25 vs. 35 ms; see in “Materials and methods”) augmented the hypointense signal and by comparison of images gave a reliable sign of USPIO presence in the tissue. A number of long-term survival postischemic rats were also tested with anti-CD8 antibodies for the cytotoxic T cells. No apparent signal was observed in five rats tested 9 months after insult, and 4 months later, on two of them the test was repeated but still there was no CD8 signal. The latter two animals were also tested after 1 week with anti-CD4 antibodies giving a positive signal as explained above.

MRI of inflammatory markers in brains of long-term survival postischemic rats. a and b Lymphocyte infiltrations monitored with USPIO antibodies against CD4 (upper rows in a and b) or CD8 receptors (bottom row in a) as compared to postischemic (null, middle row in a and b) or control (ctrl, bottom row in b) animals without USPIO—all imaged with three MRI protocols: 1 T1W, 2 T2W and 3 T2*W (the arrows indicate the hypointense regions). c Difference in images before and after Gd-contrast in postischemic animals (plates column Isch 9 months after reperfusion—top two images and 13 months bottom image) as compared to sham operated animals of similar age (plates column Ctrl). d Dilation in the retrosplenial area (encircled with white) of postischemic animals (Isch) as compared to controls (Ctrl) revealed with two MRI protocols (T1W and T2W). e Immunocytochemistry of T cells in brain parenchyma. Staining for the lymphocyte marker TCR (green) and blood vessel endothelial cells CD31 (red) in CA1 hippocampal region and striatum. Ctrl control animal, Isch postischemic animal (ortho-projections with position lines taken from Zeiss LSM 510 software); The scale bar represents 20 μm. The arrows point to intraparenchymal T cells that have crossed the BBB
In order to get further proof of T-cell infiltration in brain parenchyma of MRI-scanned postischemic animals, the brain slices of killed animals were immunostained with the lymphocyte marker TCR. Indeed, this tissue staining revealed the presence of T lymphocytes in brain parenchyma of the hippocampus and striatum, apparently outside the blood vessels stained with endothelial marker CD31 (Fig. 1e, arrows). The distribution of lymphocytes can be better seen in animated 3D z-stack reconstruction in the Online Resources 1 and 2. Some cross reaction was also seen of CD31 staining with TCR-positive lymphocytes (Fig. 1e, arrows in CA1 Isch). It was also interesting to note that the intensity of the CD31 endothelial staining was much more apparent in the postischemic than sham-control animals. This indicates that angiogenic phenomena are still present in this long-term survival postischemic model.
The above observations should point to a general phenomenon in this chronic postischemic model—persistent surveillance of inflammatory cells in particular areas of the brain tissue. It may be that this immune cell infiltration was facilitated by a compromised BBB as recently indicated by the histological data (Pluta 2006, 2007). Thus, the revealed CD4 MRI signal and TCR immunostaining prompted the testing of the BBB as the possible pathway for immune cells. This was performed by MRI with the Gd-based contrast. In fact, it was thus observed that Gd leaks through the BBB in five out of seven tested animals (Fig. 1c). On the other hand, the Gd signal was not observed in any of the control rats. The point of the BBB leakage could be ascribed to the dorsal hippocampus in the same interbrain region where the CD4 T2* USPIO signal was observed, as well as in some regions of the brainstem (Fig. 1c). This finding thus revealed probable pathways of immune cell infiltration.
Another finding that was congruent with the BBB compromise especially in the brainstem region was the observed dilation of the retrosplenial area above the pons at the frontal cerebellar level (Fig. 1d).
Immunocytochemistry of microglial markers
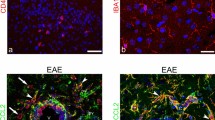
Immunocytochemistry was performed on animals killed 13 months after 10 min of global ischemia induced by cardiac arrest. Microglial markers OX42 (activated microglia) and ED1 (microglia/macrophages) were used. This study revealed significant microglial activation and infiltration in the rat hippocampus and striatum, with indications of activation in thalamic lateral dorsal nuclei and SVZ. Microglia (OX42-positive) of the ramified type was widespread in the hippocampal tissue as compared to its rare appearances in sham controls (Fig. 2a). On the other hand, ED1-positive macrophages were found in the striatum, but they were only scarce in the hippocampus (Fig. 2b) as also confirmed by the quantitative analysis (Fig. 2e). However, in the CA1 and CA3 hippocampal regions OX42- and ED1-positive granules were observed in neuronal cell bodies, suggesting some exchange of cellular material, i.e., cellular fusion between microglia/macrophages and neurons (Fig. 2a,b—CA1 and CA3). The striatum and the thalamic nuclei in fact revealed the appearance of reactive microglia of ameboid type (Fig. 2a—STR and LD, respectively). In addition, quantitative analysis of pixel intensities has confirmed a significantly higher expression of the OX42 microglial marker in all the studied structures (Fig. 2d).

Confocal images of microglial/macrophages interaction with neurons in the ischemic rat brain. Double immunofluorescence labeling for fluorescent Nissl-NeuroTrace (green) and a OX42 or b ED-1 (red) in different regions of the brain: hippocampus (CA1 and CA3), striatum (STR), thalamic lateral dorsal nuclei (LD) and subventricular zone (SVZ); c Schematic representations of frontal coronal half-sections of the anterior part of the rat brain with imaged areas marked as indicated; Ctrl control animal, Isch postischemic animal. Arrows indicate examples of labeled microglial cells. The scale bar represents 50 μm. d The ratio of OX42 over neuronal Nissl signal intensities was 1.028 ± 0.080 (CA1), 1.134 ± 0.032 (CA3), 0.527 ± 0.083 (STR), 0.489 ± 0.016 (LD), and 0.753 ± 0.079 (SVZ) in control animals (n = 6), and 1.477 ± 0.045, 1.673 ± 0.134, 1.169 ± 0.049, 0.649 ± 0.028, and 1.287 ± 0.111 in ischemic animals (n = 6), respectively. e The ratio of ED1 over neuronal Nissl signal intensities was 0.575 ± 0.142 (CA1), 0.548 ± 0123 (CA3), and 0.534 ± 0.020 (STR) in control animals (n = 6), and 0.682 ± 0.127, 0.805 ± 0.055, and 0.956 ± 0.079 in ischemic animals (n = 6), respectively. Values are presented as mean ± SEM. *P < 0.05, **P < 0.01, Student’s t test
Immunocytochemistry of neurogenesis markers
Immunocytochemistry of the ischemic animals at the SVZ (striatal level) with NeuroTrace revealed a complex structure of multilayered cells intercalated with OX42-positive ramified microglia (Fig. 2a—SVZ). This finding prompted us to study these cells more closely in order to check for the possible markers of progressive neurogenesis. Indeed, as expected, immunostaining for Ki67 and DCX revealed the presence of a significant proliferation of the progenitor stem cells in the SVZ (Fig. 3a–d). In addition, in the ischemic rats (n = 3/4) as opposed to the sham control (n = 0/4), a migratory path of DCX-positive cells out of the SVZ was revealed, suggesting that these neuroblasts may still migrate to replace damaged cells in the areas affected by neuronal degeneration (Fig. 3e). The dentate subgranular zone of the hippocampus was also checked for the neurogenesis marker DCX; however, even the use of a twice concentrated primary antibody for staining as compared to SVZ application revealed apparent staining neither in the sham-treated (n = 4) nor in the postischemic (n = 4) rats (not shown).

Confocal images of neurogenesis in the SVZ in the adult ischemic rat brain. Immunolabeling with anti-DCX (a and e) and anti-Ki67 (c) antibodies (red) and fluorescent Nissl-NeuroTrace (green); The ratio of DCX (red) signal over neuronal Nissl (green) signal intensities was 0.294 ± 0.066 in control animals and 0.546 ± 0.072 in ischemic animals (b), while the similar ratio for Ki67 over Nissl was 0.154 ± 0.1 in control animals and 0.603 ± 0.076 in ischemic animals (d) in SVZ of ischemic (n = 4) vs. control (n = 4) animals 1 year after ischemia. Values are presented as mean ± SEM. *P < 0.05, ***P < 0.001, Student’s t test; e Immunocytochemical detection of neuroblast migration from the SVZ. The arrows depict migratory neuroblasts. Neuroblast migration is absent in the control animals. f Schematic representation of frontal coronal half-section of the anterior part of the rat brain with imaged areas marked as indicated; Ctrl control animal, Isch postischemic animal, LV lateral ventricle, MS migratory stream. The scale bar represents 50 μm
Discussion
The main finding of the present study is that as late as 1 year after the global ischemic episode/reperfusion the neuroinflammatory processes are still occurring (“secondary inflammation”) as evidenced by (1) circulating USPIO-tagged CD4+ cell infiltration into brain parenchyma, (2) immunocytochemistry of infiltrated lymphocytes with the marker TCR, (3) microglial activation as indicated by OX42/ED1 markers, (4) BBB leakage of Gd-based MRI contrast, and (5) angiogenic processes as revealed by intense endothelial CD31 staining.
It is well known that global cerebral ischemia induces a profound inflammatory response involving granulocytes, T cells and at a later stage macrophages and microglial cells (Schroeter et al. 1994; Stoll et al. 1998). Among these cells particularly T cells are known to exert a dual role—an early one during infarct development (Yilmaz et al. 2006) and a late one during the adaptive processes (Hallenbeck et al. 2006). In the present study it was, however, of particular interest to note that even several months up to more than 1 year after reperfusion, there are still USPIO-traced circulating T cells that appear in the brain parenchyma. These were T cells of the CD4+ helper type. Although it cannot be excluded that the cytotoxic CD8+ lymphocytes have been already primed and invaded the brain tissue in the earlier postischemic acute period or may still be quiescent (Ridge et al. 1998), it is the circulating T helper cells that are still surveying the brain tissue months after the ischemia/reperfusion episode. These lymphocytes may do this in a facilitated manner through a definitely hampered BBB in a specific area of the dorsal hippocampus. In the same brain section but in the ventro-lateral direction, the area occupied by CD4+ T cells was also revealed. This area mainly corresponded to the caudoputamen (striatum) which in addition to the hippocampal CA1 region, is known to be a vulnerable brain region prone to selective postischemic neuronal damage (Lipton 1999). Moreover, immunocytochemical findings underlined the persistence of inflammatory cell markers, OX42-positive reactive microglia, and TCR-positive cells with enhanced endothelial staining in the striatum and hippocampus (Figs. 1e, 2a, b—CA1, CA3, STR). In addition, it is the helper (CD4+) T cells that are suggested to convey the information on a homeostatic breach through the cognate recognition of a tissue-specific self-antigen (Schwartz et al. 2009). However, caution has to be expressed as to the origin of USPIO-CD4+ cells, since it was demonstrated that following injury to the CNS and damage to the BBB, cells with the morphology of microglia were found to express the CD4 receptor (Perry and Gordon 1987). The latter cannot be excluded since there was evidence of BBB compromise (MRI with Gd-based contrast), as well as of the presence of reactive microglia (OX42 immunocytochemistry). Nevertheless, the immunocytochemical data with TCR-positive staining (Fig. 1e) support the T cell origin of the MRI signal.
The leakage of the BBB in this long-term survival ischemic rat model confirmed some previous histological data (Pluta 2006, 2007) and identified a particular region of the dorsal hippocampus as the most susceptible to inflammatory phenomena and β-amyloid peptide accumulation (Pluta et al. 2010). This region could also serve as a possible pathway for the infiltration of lymphocytes. Nevertheless, it may not be that a compromised BBB is the prerequisite for the lymphocyte infiltration into CNS. In fact, it has been shown in the studies on experimental autoimmune encephalomyelitis (model of multiple sclerosis) with USPIO particles phagocytized by macrophages that the BBB breakdown and inflammatory cell infiltration are not necessarily linked (Dousset et al. 1999a, b; Rausch et al. 2003). This was, however, contrasted by Floris et al. (2004) who demonstrated that MRI enhancement by Gd-DTPA always preceded the USPIO-enhancement. In stroke patients that received USPIO 1 week after the symptom onset, a T2/T2*W signal loss and T1W enhancement were observed albeit heterogeneous and not related to subacute lesion volume (Saleh et al. 2004a, b; Nighoghossian et al. 2007).
Immunocytochemistry on brain slices from this model has revealed a widespread distribution of OX42 microglial cellular markers for ramified microglia of the downregulated inflammatory profile in the hippocampus and ameboid proinflammatory type in subcortical regions (thalamus and striatum). However, intracellular granulations of the microglial origin were also seen in fluorescent Nissl-NeuroTrace stained hippocampal pyramidal neurons. The latter could indicate traces of cell fusion that occurred in the previous postischemic phases. It may, however, seem unusual to assume a phagocytosis-like fusion role of neurons toward microglia. Nevertheless, different phagocytotic manifestations were observed in neurons (Bowen et al. 2007), and interestingly, traces of apoptotic T cells were also found inside the perikaryon of facial motor neurons (Flügel et al. 2000). In addition, unlike OX42-positive microglia, in the present study ED1-positive macrophages were not easily identified. These observations are congruent with substantial pathologically induced intrinsic proliferation of parenchymal microglia and recruitment of monocytes (Ekdahl et al. 2009). It is also possible that the early detrimental action of microglia after acute ischemia-induced injury (e.g., causing phagocytosis and cell fusion) has been converted into a supportive state during the chronic phase in this long-term survival postischemic rat model. In fact, a beneficial, instructive role of microglia in adult neurogenesis was ascribed to the chronic postischemic phase and accordingly neurogenesis was observed in the SVZ at least 1 year after the insult (Kokaia et al. 2006; Thored et al. 2006). This is consistent with our observation of a multilayered SVZ structure of proliferating Ki67-positive cells with the notable presence of intercalated microglia, 1 year after global ischemia/reperfusion. As also shown here, this SVZ continues to produce new DCX-positive neuroblasts that were shown by Kokaia et al. (2006) to migrate into the areas affected by neuronal degeneration and then adopt a mature neuronal phenotype. Thus, neural stem cells in the SVZ are a constant source of cellular raw material, which could be used for self-repair in the brain during the recovery phase after ischemic stroke. It has been proposed that CNS-specific T cells can instruct resident microglia and thus promote progenitor proliferation in the subgranular hippocampal zone and SVZ (Ziv et al. 2006, Schwartz et al. 2009). Interestingly, it was also shown that CD4+ but not CD8+ T-lymphocytes promote adult neurogenesis in the hippocampus (Wolf et al. 2009; Huang et al. 2010). In line with the latter, our USPIO-enhanced MRI supported by TCR immunocytochemistry indicates the presence of CD4+ but not of CD8+ T cells in brain parenchyma, thus setting the stage for the cooperation of immune cells in the neurogenic niche even 1 year after the ischemic insult. However, it is worth noting that contrary to the SVZ and as supported by Darsalia et al. (2005) we have not noticed an ongoing neurogenesis in the subgranular zone of the hippocampus in this postischemic model. In fact, even though neurogenesis followed by migration of periventricular cells was observed in the hippocampus weeks after ischemia (Nakatomi et al. 2002), there were also important concerns raised about the fate of the proliferative cells in hypoxia–ischemia (Kuan et al. 2004).
It was thus evidenced that up to 1 year after the ischemia/reperfusion, insult hallmarks of neuroinflammation, a hampered BBB that facilitates T cell infiltration and activated resident microglia, are still apparent. At this stage, monocytes/macrophages are not observed in brain parenchyma. Instead, ED1-positive granules may represent traces of possible cell fusion of macrophages with pyramidal neurons. Neurogenesis in the SVZ and cell migration is still occurring; however, the fate of these cells, their phenotype and migratory path needs to be revealed. The persistent leakage of the BBB may eventually overcome cell survival strategies and thus lead to β-amyloid deposits (Pluta et al. 2009, 2010) tau pathology (Yang and Simpkins 2007) and pro-apoptotic signaling causing neurodegeneration of full-blown Alzheimer’s type.
Abbreviations
- BBB:
-
Blood–brain barrier
- CLIO:
-
Cross-linked iron oxide
- CNS:
-
Central nervous system
- DCX:
-
Doublecortin
- FITC-streptavidin:
-
Streptavidin labeled with fluorescein isothiocyanate
- FoW:
-
Field of view
- Gd-DTPA:
-
Gadolinium-diethylenetriaminepentaacetic acid
- MRI:
-
Magnetic resonance imaging
- PECAM:
-
Platelet endothelial cell adhesion molecule
- RF:
-
Radio frequency
- SPIO:
-
Small paramagnetic iron oxide particles
- SVZ:
-
Subventricular zone
- T1:
-
Longitudinal relaxation time
- T1W:
-
T1-weighted relaxation
- T2*:
-
Transversal relaxation time-“star” protocol
- T2*W:
-
T2*-weighted relaxation
- TCR:
-
T cell receptor
- TE:
-
Time to echo
- TR:
-
Repetition time
- TSE:
-
Turbo spin echo protocol
- USPIO:
-
Ultra small paramagnetic iron oxide particles
References
Arshavsky YI (2010) Why Alzheimer’s disease starts with a memory impairment: neurophysiological insight. J Alzheimer’s Dis 20:5–16
Bowen S, Ateh DD, Deinhardt K et al (2007) The phagocytic capacity of neurones. Eur J Neurosci 25:2947–2955
Darsalia V, Heldmann U, Lindvall O et al (2005) Stroke-induced neurogenesis in aged brain. Stroke 36:790–1795
Dousset V, Ballarino L, Delalande C et al (1999a) Comparison of ultrasmall particles of iron oxide (USPIO)-enhanced T2-weighted, conventional T2-weighted, and gadolinium-enhanced T1-weighted MR images in rats with experimental autoimmune encephalomyelitis. Am J Neuroradiol 20:223–227
Dousset V, Delalande C, Ballarino L et al (1999b) In vivo macrophage activity imaging in the central nervous system detected by magnetic resonance. Magn Reson Med 41:329–333
Ekdahl CT, Kokaia Z, Lindvall O (2009) Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience 158:1021–1029
Floris S, Blezer ELA, Schreibelt G et al (2004) Blood-brain barrier permeability and monocyte infiltration in experimental allergic encephalomyelitis: a quantitative MRI study. Brain 127:616–627
Flügel A, Schwaiger FW, Neumann H et al (2000) Neuronal FasL induces cell death of encephalitogenic T lymphocytes. Brain Pathol 10:353–364
Hallenbeck J, del Zoppo G, Jacobs T, Immunomodulation Workshop Participants et al (2006) Immunomodulation strategies for preventing vascular disease of the brain and heart. Stroke 37:3035–3042
Huang G-J, Smith AL, Gray DHD et al (2010) A genetic and functional relationship between T cells and cellular proliferation in the adult hippocampus. PLoS Biol 8:e1000561
Kiryk A, Pluta R, Figiel I et al (2011) Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav Brain Res 219:1–7
Kokaia Z, Thored P, Arvidsson A et al (2006) Regulation of stroke-induced neurogenesis in adult brain: recent scientific progress. Cereb Cortex 16(Suppl 1):i162–i167
Kuan CY, Schloemer AJ, Lu A et al (2004) Hypoxia–ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci 24:10763–10772
Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79:1431–1568
Nakatomi H, Kuriu T, Okabe S et al (2002) Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 110:429–441
Nighoghossian N, Wiart M, Cakmak S et al (2007) Inflammatory response after ischemic stroke: a USPIO-enhanced MRI study in patients. Stroke 38:303–307
Perry VH, Gordon S (1987) Modulation of the CD4 antigen on macrophages and microglia in rat brain. J Exp Med 165:1218–1223
Pirko I, Ciric B, Johnson AJ et al (2003) Magnetic resonance imaging of immune cells in inflammation of central nervous system. Croat Med J 44:463–468
Pluta R (2006) Is the ischemic blood-brain barrier insufficiency responsible for full-blown Alzheimer’s disease? Neurol Res 28:665–671
Pluta R (2007) Role of ischemic blood-brain barrier on amyloid plaques development in Alzheimer’s disease brain. Curr Neurovasc Res 4:121–129
Pluta R, Lossinsky AS, Mossakowski MJ et al (1991) Reassessment of a new model of complete cerebral ischemia in rats. Method of induction of clinical death, pathophysiology and cerebrovascular pathology. Acta Neuropathol 83:1–11
Pluta R, Ułamek M, Jabłoński M (2009) Alzheimer’s mechanisms in ischemic brain degeneration. Anat Rec 292:1863–1881
Pluta R, Januszewski S, Jabłoński M et al (2010) Factors in creepy delayed neuronal death in hippocampus following brain ischemia-reperfusion injury with long-term survival. Acta Neurochir Suppl 106:37–41
Rausch M, Hiestand P, Baumann D et al (2003) MRI-based monitoring of inflammation and tissue damage in acute and chronic relapsing EAE. Magn Reson Med 50:309–314
Ridge JP, Di Rosa F, Matzinger P (1998) A conditioned dendritic cell can be a temporal bridge between a CD4 T-helper and a T-killer cell. Nature 393:474–478
Saleh A, Schroeter M, Jonkmanns C et al (2004a) In vivo MRI of brain inflammation in human ischemic stroke. Brain 127:1670–1677
Saleh A, Wiedermannn D, Schroeter M et al (2004b) Central nervous system inflammatory response after cerebral infraction as detected by magnetic resonance imaging. NMR Biomed 17:63–69
Sastre M, Dewachter I, Landreth GE et al (2003) Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci 23:9796–9804
Schroeter M, Jander S, Witte OW et al (1994) Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J Neuroimmunol 55:195–203
Schwartz M, London A, Shechter R (2009) Boosting T-cell immunity as a therapeutic approach for neurodegenerative conditions: the role of innate immunity. Neuroscience 158:1133–1142
Selakovic V, Raicevic R, Radenovic L (2009) Temporal patterns of soluble adhesion molecules in cerebrospinal fluid and plasma in patients with the acute brain infraction. Dis Markers 26:65–74
Stoll G, Bendszus M (2009) Imaging of inflammation in the peripheral and central nervous system by magnetic resonance imaging. Neuroscience 158:1151–1160
Stoll G, Jander S, Schroeter M (1998) Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol 56:49–71
Sugawara T, Fujimura M, Noshita N et al (2004) Neuronal death/survival signaling pathways in cerebral ischemia. NeuroRx 1:17–25
Thored P, Arvidsson A, Cacci E et al (2006) Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells 24:739–747
Touzani O, Boutin H, LeFeuvre R et al (2002) Interleukin-1 influences ischemic brain damage in the mouse independently of the interleukin-1 type I receptor. J Neurosci 22:38–43
Wolf SA, Steiner B, Akpinarli A et al (2009) CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol 182:3979–3984
Yang SH, Simpkins JW (2007) Ischemia-reperfusion promotes tau and beta-amyloid pathology and a progressive cognitive impairment. In: Pluta R (ed) Ischemia-reperfusion pathways in Alzheimer’s disease. Nova Science Publishers, Inc., New York, pp 113–138
Yilmaz G, Arumugam TV, Stokes KY et al (2006) Role of T lymphocytes and interferon-γ in ischemic stroke. Circulation 113:2105–2112
Ziv Y, Ron N, Butovsky O et al (2006) Immune cells contribute to maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci 9:268–275
Acknowledgments
The authors are grateful to Dr. Goran Bačić for his continuous expert support in designing and analyzing MRI experiments. This study was supported by grant of Ministry of Science and Technological Development Republic of Serbia (Grant No. 41005), funds from Mossakowski Medical Research Centre (T5), Polish Ministry of Science and Higher Education (2007–2010-Cost/253/2006), and ESF-COST Action B30.
Author information
Authors and Affiliations
Corresponding author
Additional information
R. Pluta and P. R. Andjus contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
429_2011_336_MOESM1_ESM.mpg
Supplementary material 1 Online Resource 1. Animated reconstruction of the Z-stack of the image in Fig. 1e for the postischemic CA1 hippocampal region (optical slice thickness – 0.79 μm, number of slices – 52, stack size – 40.2 μm) (MPEG 621 kb)
429_2011_336_MOESM2_ESM.mpg
Supplementary material 2 Online Resource 2. Animated reconstruction of the Z-stack of the image in Fig. 1e for the postischemic striatal region (optical slice thickness – 1.99 μm, number of slices – 14, stack size – 25.9 μm) (MPEG 469 kb)
Rights and permissions
About this article
Cite this article
Sekeljic, V., Bataveljic, D., Stamenkovic, S. et al. Cellular markers of neuroinflammation and neurogenesis after ischemic brain injury in the long-term survival rat model. Brain Struct Funct 217, 411–420 (2012). https://doi.org/10.1007/s00429-011-0336-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00429-011-0336-7