
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that has been characterized by gross cortical atrophy, cellular neurodegeneration, reactive gliosis, and the presence of microscopic extracellular amyloid plaques and intracellular neurofibrillary tangles. Earlier diagnoses of AD would be in the best interest of managing the patient and would allow for earlier therapeutic intervention. By measuring the apparent diffusion coefficient (ADC) using diffusion-weighted imaging (DWI), a type of magnetic resonance imaging (MRI), one can quantify alterations in water diffusivity resulting from microscopic structural changes in the cell at early stages that are associated with pathophysiological processes of brain injury and/or disease progression. Whether or not this methodology is useful for AD is a question under examination. For example, DWI in suspected AD patients has shown increases in mean ADC values in the hippocampus and diminished diffusion anisotropy in the posterior white matter. However, in some cases, hippocampal ADC values appear not to change in AD patients. Moreover, to our knowledge, all DWI studies in suspected AD patients to date are technically incomplete in experimental design, because corresponding histological sections demonstrating actual plaque deposition are lacking and so it is not clear that ADC changes actually correspond to plaque deposition. In our study, we used DWI in the TgCRND8 transgenic model of Alzheimer’s disease in conjunction with histological techniques and found robust plaque deposition in the transgenic strain in older animals (12–16 months old). However, we did not find statistically significant changes (p > 0.05) in ADC values (although ADC values in TgCRND8 mice did decrease in all regions examined) in mice 12–16 months old. Collectively, recent results from human studies and in rodent AD transgenic models support our findings and suggest that amyloid beta plaque load is not likely the major or primary component contributing to diffusional changes, if they occur.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
At the present time, physicians currently lack standard procedures and reliable markers by which to classify and/or diagnose Alzheimer’s disease (AD). In fact, clinicians currently can only make a provisional diagnosis of probable AD using neuropsychological testing, and direct pathologic examination of brain tissue at autopsy remains the only definitive method for establishing a diagnosis of AD. Typically, autopsy data show neurofibrillary tangles, amyloid plaques, and atrophy, which occur first in the entorhinal cortex and hippocampal formation before progressing to other brain regions (Fox et al. 1996). Earlier diagnoses of AD, on the other hand, would be in the best interest of managing the patient and would allow for either earlier therapeutic intervention or additional therapeutic options once more effective AD drugs become available.
Diffusion-weighted imaging (DWI), a quantitative magnetic resonance imaging (MRI) technique, is sensitive to water diffusion in tissue caused by the random motion of water molecules (Bammer 2003). DWI has been used for imaging CNS disorders previously (e.g., ischemia, head trauma) (Hanstock et al. 1994; Warach et al. 1995; Assaf et al. 1997; Albensi et al. 2000; Huisman 2003) and might also allow earlier detection of AD (Bozzao et al. 2001). Past studies have confirmed that DWI permits earlier detection of acute brain injury compared with T2-weighted imaging due to increased sensitivity (Bammer 2003). In the context of acute brain injury, DWI has been shown to be very sensitive to diffusion restriction and cytotoxic edema, whereas T2-weighted imaging is very sensitive to vasogenic forms of edema and free extracellular water (Radaideh et al. 2003). By measuring the apparent diffusion coefficient (ADC) with DWI one can quantify alterations in apparent water diffusivity resulting from microscopic structural changes in the cell that are associated with brain injury and/or disease progression. In particular, the ADC map has been reported to more accurately predict restricted diffusion as it represents the distribution of ADCs throughout the brain without the influence of the T2 contrast (Radaideh et al. 2003).
When brain structures, such as the hippocampal formation, are pathologically disrupted as a result of neurodegeneration, ADC values might change accordingly in conjunction with alterations in extracellular and intracellular microstructure due to amyloid deposition, neuritic degeneration and/or cytoskeletal breakdown. Microstructures in the CNS normally restrict water molecular diffusion and so pathological processes in AD that alter tissue integrity might remove some of the restrictive barriers resulting in increased measured ADCs. In support of this notion, one past study reported that patients with the syndrome of mild cognitive impairment (MCI), a transitory state between normal cognition and AD (Kantarci et al. 2005), had hippocampal ADCs significantly increased, whereas normal individuals without MCI did not have elevated ADCs (Kantarci et al. 2001). DWI studies in suspected AD patients to date have in some cases also shown significantly higher mean diffusivity in the cingulate, temporal, and white matter when compared with control patients (Sandson et al. 1999; Kantarci et al. 2001, 2002, 2005). Furthermore, DWI in suspected AD patients have shown increases in mean ADC values in the hippocampus and diminished diffusion anisotropy in the posterior white matter (Sandson et al. 1999; Kantarci et al. 2001, 2002, 2005).
While these data are interesting, these studies suffer from several unresolved issues. First, not all results thus far have been in agreement and in some cases, hippocampal ADC values appear not to change in AD patients (Hanyu et al. 1998). Secondly, most DWI studies (Hanyu et al. 1998; Yoshiura et al. 2003; Kantarci et al. 2005) in suspected AD patients lack corresponding histological sections demonstrating actual plaque deposition. Consequently, it is not clear if and how ADC changes actually correspond to plaque deposition.
To address some of these outstanding questions, we chose to use the TgCRND8 strain, a standard transgenic mouse model of Alzheimer’s disease. Normally, mice do not develop plaques, however, TgCRND8 mice have been engineered to possess a double mutant form of the human APP695 gene with both the Swedish and Indiana mutations under the control of the hamster Prion P promotor. It is well documented that these mutations produce accelerated amyloid deposition and cognitive deficits in animals after 3 months of age (Kobayashi and Chen 2005; Rak et al. 2007). An animal model of this type permits evaluations of amyloid plaque deposition and other anatomical changes associated with AD pathology. In this study, our objectives were to evaluate anatomical and physiological brain changes in the AD mouse model as a result of disease progression over a 16-month period, and to compare ADC values obtained in brain regions suspected to be involved in AD-like pathogenesis with corresponding histological sections demonstrating plaque deposition.
Methods
Animal model
All animal studies were approved by the University of Manitoba Protocol Management and Review Committee. Young, pre-plaque (3 months old) versus older control and TgCRND8 animals (12–16 months) were used for this study.
Thirteen mice (12–16 months old) were imaged with diffusion-weighted MRI of which 11 datasets were usable (4 female wild-type and 7 male TgCRND8). Twelve mice (six 13-months old and six 3-months old) were imaged with high-resolution T2-weighted imaging (3 WT and 3 TgCRND8 in each age group).
TgCRND8 and littermate control mice were originally a gift from Dr. Westaway who engineered these mice at the Centre for Research in Neurodegenerative Diseases (CRND), University of Toronto, and who previously described this strain in detail (Chishti et al. 2001). Once brought to our institution, the colony was bred for supplying additional animals. The TgCRND8 strain is a model for Alzheimer’s disease-like amyloid pathogenesis that expresses a double mutant form of human amyloid precursor protein (APP) 695 (K670N/M671L and V717F). For this study, we used male TgCRND8 (+/−) mice and their female non-Tg littermates (−/−); to our knowledge no differences between gender have been reported in TgCRND8 mice with regard to general AD pathology, plaque deposition, or behavior. All mice were housed independently and maintained on ad libitum food and water with a 12-h light/dark cycle.
Animal preparation for scanning
Mice were anesthetized with isoflurane (5% for induction and 1.5–2% for maintenance) in oxygen and nitrous oxide (O2:N2O = 30:70) for the duration of the scans. The inflow line of the inhalant and outflow of the exhaust were connected to a nose cone. A scavenging line removed excess isoflurane from the magnet bore. Respiration rates were monitored using a standard monitoring and gating system (Model 1025, SA Instruments, Inc., Stony Brook, NY, USA). The magnet bore temperature was monitored using an extra-flexible PVC-insulated probe with a PVC-coated tip thermocouple (Cole-Parmer Canada, Anjou, QC, Canada) and maintained between 33 and 34°C using warm air flowing into the magnet bore. The animals rested in a prone position in a custom-made polyethylene cradle with the head placed in a 2.4-cm inner diameter, modified quadrature birdcage volume coil (NRC-IBD, Winnipeg, MB, Canada). Imaging was performed on a 7 Tesla (T) horizontal Bruker magnet and Biospec/3 console (BioSpin GmbH, Ettlingen, Germany) running Paravision 2.0.1 acquisition software.
Image acquisition
Pilot FLASH scans were performed to correctly position the animal in the magnet. Once proper placement was achieved, two sets of coronal plane images were acquired with a focus on the hippocampus.
Diffusion-weighted images were acquired in 12–16 months old mice using a magnetization prepared TurboFLASH imaging sequence (Thomas et al. 1998). Two of the control mice (12–16 months old) and one of the transgenic mice (12–16 months old) were imaged with a 30 × 30-mm field of view (FOV), with the remaining eight mice (12–16 months old) imaged with a 40 × 40-mm FOV to resolve heating problems in the gradient coils. Diffusion-weighted images were acquired in three directions (x, y, and z) with a 128 × 128 matrix size, in-plane resolution of 234 × 234 μm or 313 × 313 μm (dependent on FOV), a slice thickness of 1 mm, and 32 averages. Eight diffusion-weighted images were acquired in each direction with varying gradient strengths (gradient pulse duration, δ = 18 ms; gradient pulse separation, Δ = 20 ms; gradient pulse strength, g = 8.0, 16.1, 24.11, 32.2, 40.2, 48.3, 56.3, and 64.4 mT/m) with respective b values of 21.0, 84.1, 189.1, 336.3, 525.4, 756.6, 1,029.8, and 1,345.0 s/mm2 calculated using the diffusion-weighting factor equation for symmetric and square gradient pulses, b = γ2 g 2δ2(Δ − δ/3) (Stejskal and Tanner 1965). Distortions due to the phase shift are corrected for in the quadratic summation of two complementary images at each b value, with the second image having a 90° flip-back pulse phase shifted by π/2 with respect to the first (Thomas et al. 1998). Scan time was 16 min and 20 s for each set of diffusion-weighted images, for a total DWI scan time of 3 × 2 × 16 min or 1 h 38 min.
High resolution T2-weighted images were acquired in six 3-months old (3 WT & 3 TgCRND8) and six 12–16 months old (3 WT & 3 TgCRND8) mice using a multi-slice single-echo sequence (MSSE) with five slices 1 mm apart, TR 1540 ms, TE 80 ms, 25 × 25 mm FOV, 256 × 256 matrix size, 98 × 98 μm in-plane resolution, a slice thickness of 0.75 mm, and 16 averages with a total scan time of 1 h 45 m.
Image and data analysis
Once MR images were obtained they were further analyzed by two different methods. The high resolution T2-weighted images were examined for abnormalities in the hippocampal and neocortical regions. ADC maps were calculated for each diffusion direction (ADCx, ADCy, and ADCz) as well as the mean ADC [(ADCx + ADCy + ADCz)/3] in MATLAB using linear regression analysis of the diffusion-weighted MR signal:
where S 0 is the signal intensity when no diffusion-weighted gradients are applied (b = 0).
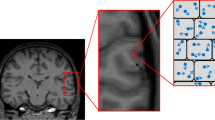
Regions of interest were selected using MATLAB in the low b value (b = 21.0 s/mm2) x-direction diffusion-weighted image. Regions of interest included the left and right regions of the hippocampus, left and right regions in the cerebral cortex, the left and right hemispheres of the brain, and the third ventricle (Fig. 1). Proper identification of each region was made with the aid of SHIVA visualization software and the MAP mouse brain atlas (MacKenzie-Graham et al. 2004) (http://www.loni.ucla.edu/MAP/). An average ADC value was calculated for each ROI using MATLAB. These data were statistically analyzed between the TgCRND8 and non-Tg mice using a two-tailed t test.

Sample region of interest (ROI) determination for ADC calculations in the hippocampal formation–coronal plane of mouse brain. Diffusion-weighted image (b = 21 s/mm2, x-direction) used for selection ROIs. The red and blue outlined regions show the left/right hippocampus and the yellow and green regions represent the left/right regions in the cerebral cortex, respectively. The cyan outlined region contains the third ventricle and the white regions are the left and right hemispheres of the brain. Scale bar 1 mm
Histology
Following the completion of MRI scanning, mice were maintained under deep isoflurane anesthetization via nose cone inhalation and perfused with formalin intracardially. For this procedure, mice were positioned supine with limbs outstretched. An intrathoracic incision was subsequently made to expose the heart, where the right atrium was nicked, and formalin was then injected into the left ventricle. Sufficient formalin was used to replace all blood. The mice were then decapitated, and the brains removed. Once removed, the brains were fixed and paraffin embedded. Coronal slices spanning the hippocampus were prepared using a thickness of 6 μ. Samples were dewaxed with xylene and rehydrated by using successively decreasing percentages of ethanol. Amyloid beta plaque deposition was stained with the indicator Congo Red (Accustain Kit; Sigma-Aldrich, USA) and the nucleus was stained with the indicator DAPI (Calbiochem, USA). Staining with DAPI enabled us to clearly identify the granule cell layer in the hippocampus for image processing. Staining with Congo Red allowed us to directly visualize amyloid plaque deposition. Samples were imaged using a fluorescent microscope with a DAPI filter at 323 nms and a Texas Red filter at 520 nms. Images were then overlaid in order to visualize cell structure and plaque characteristics.
Results
High resolution T2-weighted images
The high resolution T2-weighted images were visualized with MATLAB. Overall anatomical features were evaluated and compared in TgCRND8 versus control littermate strains. Anatomical features at this level of observation were largely unremarkable and appeared the same between the two strains. A dark line in the CA1 region (Fig. 2) was prominent in the WT 12–16-month-old mice but completely absent in the TgCRND8 12–16 month-old mice. An ex vivo T2w MRI of one WT and one TgCRND8 confirmed this feature. The line was not as prominent in WT 3-month-old animals and was barely visible in TgCRND8 3-month-old animals. The significance of this observation is not clear. However, visual examination of corresponding histological sections showed a normal CA1 pyramidal cell body layer in both WT and TgCRND8 12–16-month old sections (Fig. 3c, d, orange arrows).

Representative images from T2-weighted coronal scans of normal CRND8 WT littermate (left) versus TgCRND8 (right) mouse brains. Red arrow highlights the CA1 pyramidal cell layer left panel, which is lost in the CRND8 Tg mouse right panel (n = 3 WT; n = 3 Tg)

Representative comparison between littermate control brain slices, WT (a, c) with TgCRND8 brain slices (b, d) obtained from animals previously scanned with MRI. Slices were stained with Congo red, an agent selective for amyloid beta (Aβ). The granule cell body layer (green arrows) of the dentate gyrus subfield in the hippocampus is indicated in all panels. Only Tg slices taken from older animals (d) exhibit amyloid plaque deposition (orange spots). The orange arrows point to the CA1 pyramidal cell body layer and show no obvious difference in appearance between older WT and TgCRND8 sections. Magnification approx. 5×
DWI analysis
ADC values were calculated from 11 animals (12–16 months old) using hippocampal and neocortical ROIs in TgCRND8 versus control littermate mice and are shown in Table 1. No significant differences were found between any of the groups shown (p > 0.05).
Histology
Mice from all groups were stained for both amyloid beta plaque deposits and their nuclei. Staining with DAPI enabled us to clearly identify the granule cell layer of the dentate gyrus region of the hippocampus. Staining with Congo Red allowed visualization of plaque deposition. All mice of wild-type genotype, regardless of age, failed to produce amyloid beta plaques (Fig. 3). By 12–16 months of age, AD pathology demonstrated by plaque burden was quite severe both in terms of number of plaques and their size in TgCRND8 mice as compared to non-Tg mice.
Discussion
In this study, we found robust plaque deposition in TgCRND8 animals at 12–16 months, but not in 3-months old TgCRND8 animals or in control animals (young or old), as expected. In addition, ADC values measured in specific regions of the hippocampus and cerebral cortex showed no statistically significant change when controls were compared to TgCRND8 animals at 12–16 months of age. However, there was a slight nonsignificant ADC decrease in every brain region examined (i.e., hippocampus, cortex, whole brain, etc.) in the TgCRND8 strain in mice 12–16 months old when compared with control. MRI methodologies such as DWI and diffusion tensor imaging (DTI) are rapidly evolving and interpreting their resultant data should first begin with a firm understanding of the pathological process being evaluated.
For example, AD is a progressive neurodegenerative disorder that has been characterized by gross cortical atrophy (widened sulci and narrowed gyri), cellular neurodegeneration, reactive gliosis, and the presence of microscopic extracellular amyloid plaques and intracellular neurofibrillary tangles (Albert 1996; Chetelat and Baron 2003). Overall, the volume of the central cerebral white matter is reduced (Englund et al. 1988). In particular, AD pathology involves the cerebral cortex, including severe lesioning in the hippocampus and entorhinal cortex, and subcortical and brainstem structures (Giannakopoulos et al. 1997). Cortical lesions are associated with senile plaques of the neuritic type in the neuropil and the formation of neurofibrillary tangles in nerve cell bodies. Tangles are masses of argyrophilic fibrils that form in the cytoplasm and apical dendrites of neurons. Ultrastructural studies show that tangles are composed of mainly paired helical filaments (20 nm diameter) made of tau protein. Plaques are rounded deposits of amyloid beta (Aβ), derived from APP, varying in size from 5 to 200 μm in diameter and appearing in two types, neuritic or diffuse. However, tangles and amyloid plaques are not specific for only AD and can be found in other neurodegenerative disorders.
To evaluate AD anatomy and pathophysiology, several diffusion MRI and DTI studies have been conducted (Hanyu et al. 1998; Sandson et al. 1999; Bozzao et al. 2001; Kantarci et al. 2001, 2005; Yoshiura et al. 2003; Song et al. 2004; Sun et al. 2005; Wang and Su 2006). However, to our knowledge, in studies with humans, ADC results were not correlated with histopathology. In addition, very few DWI or DTI studies have been conducted in Alzheimer’s disease transgenic mouse models (Mueggler et al. 2004; Sun et al. 2005; Harms et al. 2006), where an evaluation of general histopathology and plaque deposition could easily be made in conjunction with diffusional changes. Of the human studies, many have shown changes in ADC values in AD, but some have not. In a study by Hanyu et al. (1997), ADCs in the anterior and posterior white matter were significantly higher in AD patients as compared to control patients. However, in another study by Bozzao et al. (2001), DWI showed no significant differences in anisotropy in posterior white matter. In another study by Hanyu et al. (1998), no significant differences in ADC values in the hippocampus between AD and control patients were detected, but changes in other regions were seen (e.g., temporal stem). On the other hand, Warach and colleagues (Sandson et al. 1999) found that ADC values in the hippocampus were increased in AD patients when compared with control. In more recent studies by Kantarci et al. (2002, 2005), significant increases in ADC values were found in the hippocampus in both MCI patients and in AD patients when compared with control patients. In an animal study by Song et al. (2004) relative anisotropy was significantly decreased while radial diffusivity and the diffusion tensor trace [Tr(D), equivalent to three times the mean ADC] were significantly increased in white matter in brains of 15-month-old PDAPP mice. In another rodent study by Sun et al. (2005) reduced Tr(D) was observed in both gray and white matter in Tg2576 mice 8–16 months old. Given these differences, we are forced to examine diffusion methodology more closely.
The basic concepts associated with diffusion MRI were introduced in the 1980s by Le Bihan and others (2001). Basser et al. (1994) formally introduced the concept of DTI into the MRI literature, which more fully characterized and exploited the effects of diffusion MRI. In DTI, the idea was introduced that diffusion is a three-dimensional process that is not adequately described by scalar measures like the ADC. In DTI, diffusion gradients are applied in several spatial directions (e.g., 6 directions) to determine a multidimensional diffusion tensor. One important application of DTI is that of brain fiber tracking, which is used for mapping white matter tracks (Le Bihan et al. 2001).
Our study and some other past studies did not utilize DTI protocols, which could be one reason why our DWI scans did not detect significant differences in diffusion between TgCRND8 mice versus their controls. High resolution DTI studies coupled with a voxel-based analysis would detect subtle changes that we are unable to detect with DWI and would thus make an interesting future study. However, most recent studies using DTI in humans and animals focus on evaluating white matter integrity. In these studies, DTI has revealed impairments in white matter in AD and MCI in some cases, while other DTI studies have not detected any differences in mean diffusivity or directional diffusion in AD or MCI patients (Stebbins and Murphy 2009). Various measures of water diffusion can be made with DWI including the overall magnitude of diffusion (diffusivity) as well as the observation that diffusion tends to be directionally dependent. This latter characteristic, however, is measured more appropriately by calculating a diffusion tensor for each voxel using DTI. In any case, the discrepancies in the above studies raise several questions including what neuropathological process(es) is (are) leading to changes in diffusional parameters in some cases and not in others.
Given that cellular swelling and inflammatory responses are pathological processes that begin in early and mid stages in AD and given the fact that histopathology was not evaluated in the above-mentioned human MRI studies, it cannot be ruled out that changes in diffusional parameters in past AD human studies were actually a reflection of inflammatory processes and not a result of plaque deposition. Of course, at the time of death, plaque deposition in humans would presumably be robust. In addition, past human studies in MCI patients showed elevated ADCs in hippocampal regions, where it would not seem likely that these patients would have significant plaque deposition at this stage, which implies that elevated ADCs are not linked to increased plaque deposition. Using a cerebral amyloidosis transgenic mouse model, Mueggler et al. (2004) demonstrated reduced ADC in neocortical areas of 25-month-old APP23 transgenic mice. They attribute these changes to extracellular deposition of fibrillar amyloid and/or associated glial proliferation and hypertrophy. Similarly, in a rodent study conducted by Sun et al. (2005) reduced Tr(D) generally coincided with amyloid plaque deposition in Tg2576 mice 8–16 months old, however, there was no statistically significant positive correlation between Aβ load and DTI changes (i.e., in hippocampus and corpus callosum) in this strain in the age range tested, which is in agreement with our findings.
There were a few limitations in our study that we should note. This study was a pilot exploratory analysis that utilized a small sample size. Although, we did not find statistically significant differences in mean ADCs between groups, a larger sample size having more power might detect differences we were not able to. In addition, we used both male and female mice in our ADC analysis as a matter of convenience and budget, where we compared males of one group to females of the other group. We saw no significant difference in mean ADC between these groups as noted above, however, we cannot completely discount the possibility that gender differences obscured an effect. Finally, by scanning at higher resolution, one would reduce the possibility of introducing partial volume effects that could mask potential differences in ADC values between strains.
In conclusion, we found robust plaque deposition in TgCRND8 animals at 12–16 months of age, which did not coincide with changes in ADC values in hippocampal or neocortical regions measured in the same animals since we found no significant difference in ADC values at 12–16 months in controls versus Tg littermates. Interestingly, the findings in our study are consistent with other AD transgenic studies using the Tg2576 AD transgenic model (Sun et al. 2005) that found no correlation between plaque load and diffusional changes. Collectively, results from human studies and in rodent AD transgenic models suggest that Aβ plaque load is not likely the major or primary component contributing to diffusional changes if and when they are seen.
References
Albensi BC, Knoblach SM, Chew BG, O’Reilly MP, Faden AI, Pekar JJ (2000) Diffusion and high resolution MRI of traumatic brain injury in rats: time course and correlation with histology. Exp Neurol 162:61–72
Albert MS (1996) Cognitive and neurobiologic markers of early Alzheimer disease. Proc Natl Acad Sci USA 93:13547–13551
Assaf Y, Beit-Yannai E, Shohami E, Berman E, Cohen Y (1997) Diffusion- and T2-weighted MRI of closed-head injury in rats: a time course study and correlation with histology. Magn Reson Imaging 15:77–85
Bammer R (2003) Basic principles of diffusion-weighted imaging. Eur J Radiol 45:169–184
Basser PJ, Mattiello J, LeBihan D (1994) MR diffusion tensor spectroscopy and imaging. Biophys J 66:259–267
Bozzao A, Floris R, Baviera ME, Apruzzese A, Simonetti G (2001) Diffusion and perfusion MR imaging in cases of Alzheimer’s disease: correlations with cortical atrophy and lesion load. AJNR Am J Neuroradiol 22:1030–1036
Chetelat G, Baron JC (2003) Early diagnosis of Alzheimer’s disease: contribution of structural neuroimaging. Neuroimage 18:525–541
Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D (2001) Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276:21562–21570
Englund E, Brun A, Alling C (1988) White matter changes in dementia of Alzheimer’s type biochemical and neuropathological correlates. Brain 111(Pt 6):1425–1439
Fox NC, Warrington EK, Freeborough PA, Hartikainen P, Kennedy AM, Stevens JM, Rossor MN (1996) Presymptomatic hippocampal atrophy in Alzheimer’s disease. A longitudinal MRI study. Brain 119(Pt 6):2001–2007
Giannakopoulos P, Hof PR, Michel JP, Guimon J, Bouras C (1997) Cerebral cortex pathology in aging and Alzheimer’s disease: a quantitative survey of large hospital-based geriatric and psychiatric cohorts. Brain Res Brain Res Rev 25:217–245
Hanstock CC, Faden AI, Bendall MR, Vink R (1994) Diffusion-weighted imaging differentiates ischemic tissue from traumatized tissue. Stroke 25:843–848
Hanyu H, Shindo H, Kakizaki D, Abe K, Iwamoto T, Takasaki M (1997) Increased water diffusion in cerebral white matter in Alzheimer’s disease. Gerontology 43:343–351
Hanyu H, Sakurai H, Iwamoto T, Takasaki M, Shindo H, Abe K (1998) Diffusion-weighted MR imaging of the hippocampus and temporal white matter in Alzheimer’s disease. J Neurol Sci 156:195–200
Harms MP, Kotyk JJ, Merchant KM (2006) Evaluation of white matter integrity in ex vivo brains of amyloid plaque-bearing APPsw transgenic mice using magnetic resonance diffusion tensor imaging. Exp Neurol 199:408–415
Huisman TA (2003) Diffusion-weighted imaging: basic concepts and application in cerebral stroke and head trauma. Eur Radiol 13:2283–2297
Kantarci K, Jack CR Jr, Xu YC, Campeau NG, O’Brien PC, Smith GE, Ivnik RJ, Boeve BF, Kokmen E, Tangalos EG, Petersen RC (2001) Mild cognitive impairment and Alzheimer disease: regional diffusivity of water. Radiology 219:101–107
Kantarci K, Xu Y, Shiung MM, O’Brien PC, Cha RH, Smith GE, Ivnik RJ, Boeve BF, Edland SD, Kokmen E, Tangalos EG, Petersen RC, Jack CR Jr (2002) Comparative diagnostic utility of different MR modalities in mild cognitive impairment and Alzheimer’s disease. Dement Geriatr Cogn Disord 14:198–207
Kantarci K, Petersen RC, Boeve BF, Knopman DS, Weigand SD, O’Brien PC, Shiung MM, Smith GE, Ivnik RJ, Tangalos EG, Jack CR Jr (2005) DWI predicts future progression to Alzheimer disease in amnestic mild cognitive impairment. Neurology 64:902–904
Kobayashi DT, Chen KS (2005) Behavioral phenotypes of amyloid-based genetically modified mouse models of Alzheimer’s disease. Genes Brain Behav 4:173–196
Le Bihan D, Mangin JF, Poupon C, Clark CA, Pappata S, Molko N, Chabriat H (2001) Diffusion tensor imaging: concepts and applications. J Magn Reson Imaging 13:534–546
MacKenzie-Graham A, Lee EF, Dinov ID, Bota M, Shattuck DW, Ruffins S, Yuan H, Konstantinidis F, Pitiot A, Ding Y, Hu G, Jacobs RE, Toga AW (2004) A multimodal, multidimensional atlas of the C57BL/6 J mouse brain. J Anat 204:93–102
Mueggler T, Meyer-Luehmann M, Rausch M, Staufenbiel M, Jucker M, Rudin M (2004) Restricted diffusion in the brain of transgenic mice with cerebral amyloidosis. Eur J Neurosci 20:811–817
Radaideh M, Devine C, Schomer D, Kumar AJ, Cacayorin ED, Song J, Lutzker S, Leeds N (2003) Correlating the basic chronological pathophysiologic neuronal changes in response to ischemia with multisequence MRI imaging. Neurographics 2(2):Article 1
Rak M, Del Bigio MR, Mai S, Westaway D, Gough K (2007) Dense-core and diffuse Abeta plaques in TgCRND8 mice studied with synchrotron FTIR microspectroscopy. Biopolymers 87:207–217
Sandson TA, Felician O, Edelman RR, Warach S (1999) Diffusion-weighted magnetic resonance imaging in Alzheimer’s disease. Dement Geriatr Cogn Disord 10:166–171
Song SK, Kim JH, Lin SJ, Brendza RP, Holtzman DM (2004) Diffusion tensor imaging detects age-dependent white matter changes in a transgenic mouse model with amyloid deposition. Neurobiol Dis 15:640–647
Stebbins GT, Murphy CM (2009) Diffusion tensor imaging in Alzheimer’s disease and mild cognitive impairment. Behav Neurol 21:39–49
Stejskal EO, Tanner JE (1965) Spin diffusion measurements: spin echoes in the presence of a time-dependent field gradient. J Chem Phys 42:288–292
Sun SW, Song SK, Harms MP, Lin SJ, Holtzman DM, Merchant KM, Kotyk JJ (2005) Detection of age-dependent brain injury in a mouse model of brain amyloidosis associated with Alzheimer’s disease using magnetic resonance diffusion tensor imaging. Exp Neurol 191:77–85
Thomas DL, Pell GS, Lythgoe MF, Gadian DG, Ordidge RJ (1998) A quantitative method for fast diffusion imaging using magnetization-prepared TurboFLASH. Magn Reson Med 39:950–960
Wang H, Su MY (2006) Regional pattern of increased water diffusivity in hippocampus and corpus callosum in mild cognitive impairment. Dement Geriatr Cogn Disord 22:223–229
Warach S, Gaa J, Siewert B, Wielopolski P, Edelman RR (1995) Acute human stroke studied by whole brain echo planar diffusion-weighted magnetic resonance imaging. Ann Neurol 37:231–241
Yoshiura T, Mihara F, Tanaka A, Ogomori K, Ohyagi Y, Taniwaki T, Yamada T, Yamasaki T, Ichimiya A, Kinukawa N, Kuwabara Y, Honda H (2003) High b value diffusion-weighted imaging is more sensitive to white matter degeneration in Alzheimer’s disease. Neuroimage 20:413–419
Acknowledgments
This study was supported with funds from the Manitoba Medical Service Foundation (MMSF), the St. Boniface General Hospital Research Foundation, the University of Manitoba, and the Manitoba Health Research Council (MHRC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Thiessen, J.D., Glazner, K.A.C., Nafez, S. et al. Histochemical visualization and diffusion MRI at 7 Tesla in the TgCRND8 transgenic model of Alzheimer’s disease. Brain Struct Funct 215, 29–36 (2010). https://doi.org/10.1007/s00429-010-0271-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00429-010-0271-z