
Abstract
MTHFR C677T and Helicobacter pylori infection are believed to play critical roles in the DNA methylation process, an epigenetic feature frequently found in gastric cancer. The aim of this study was to verify the associations between the MTHFR C677T polymorphism and the methylation status of three gastric cancer-related genes. The influence of H. pylori strains was also assessed. DNA extracted from 71 gastric tumor samples was available for MTHFR C677T genotyping by PCR-RFLP, promoter methylation identification by MS-PCR and H. pylori detection and posterior subtyping (cagA and vacA genes) by PCR. In the distal tumors, a positive correlation was found between the methylation of CDKN2A and the allele T carriers (r = 0.357; p = 0.009). Considering the eldest patients (age ≥ 60 years old), this correlation was even higher (r = 0,417; p = 0.014). H. pylori infection by highly pathogenic strains (cagA+/vacAs1m1) was also found correlated to promoter methylation of CDKN2A and the allele T carriers in distal tumors (r = 0.484; p = 0.026). No significant correlation was verified between MTHFR C677T genotype and promoter methylation status when we analyzed the general sample. DNA methylation in CDKN2A associated to the MTHFR 677T carrier is suggested to be a distal tumor characteristic, especially in those 60 years old or older, and it seems to depend on the infection by H. pylori cagA/vacAs1m1 strains.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric adenocarcinoma is one of the most frequent malignancies worldwide and it is globally the second leading cause of cancer-related deaths [1]. Brazil is one of the countries with a higher incidence, mainly in the Southeast and Northeast regions [2]. Gastric cancer has multifactorial etiology, being caused by environmental, genetic and epigenetic factors, which may interact to the tumor promotion and progression [3]. Epidemiological studies have shown an association between disturbances of folate metabolism and increased risk of gastric cancer, including low intake of folate, low levels of folate in blood or genetic factors affecting folate metabolism, such as polymorphisms in folate-metabolizing genes [4–6].
Methylenetetrahydrofolate reductase (MTHFR) acts centrally in folate metabolism, catalyzing the 5-methylenetetrahydrofolate synthesis reaction, the predominant circulatory form of folate and carbon donor for the remethylation of homocysteine to methionine. This pathway leads to the production of S-adenosylmethionine, the universal donor of methyl radical in human, which is required for DNA methylation [7]. Thus, polymorphisms that reduce the MTHFR activity could influence the status of DNA methylation and would explain individual susceptibility to gastric cancer [8]. A C/T nucleotide change at 677 position of the MTHFR gene (MTHFR C677T), resulting in an alanine-to-valine substitution in the MTHFR protein, was found to produce a thermolabile variant of the MTHFR and significantly reduces the enzyme activity [9]. Furthermore, a recent meta-analysis carried out by Boccia et al. reported a significant positive correlation between gastric cancer and the MTHFR 677TT genotype [10].
DNA methylation is an important epigenetic feature of DNA that plays critical roles in gene regulation and cellular differentiation mechanisms. Methylation of promoter, especially in the CpG islands, culminates in gene silencing. Gastric cancer is one of the human tumors with higher number of silenced genes by methylation of their CpG islands [11]. Currently, the available data about the role of MTHFR C677T polymorphism in relation to the aberrant DNA methylation in gastric cancer is inconsistent and incomplete. It has been suggested that this polymorphism plays a dual role on the methylation status of promoter regions depending on the target gene [8, 12].
Another important etiological factor in gastric cancer is Helicobacter pylori infection. Bacterial virulence factors are characteristics present in some bacteria which enable them to cause disease. These are, mainly, vacuolating cytotoxin production associated to certain types of vacA (vacuolating cytotoxin gene A) alleles and cagA (cytotoxin associated gene product A) [13]. H. pylori strains expressing cagA and more active forms of vacA, such as the s1m1 genotype, are more likely to cause disease [14].
Despite of some advances, the molecular mechanisms associated with H. pylori in gastric carcinogenesis remains poorly understood. One of the promoting effects has been attributed to the induction of inflammation and aberrant methylation. It has been reported that frequencies or levels of CpG islands methylation of certain genes correlate with H. pylori infection, histological or serological severity of gastritis, and gastric cancer occurrence [12]. The observation that H. pylori-induced methylation is reversed after H. pylori eradication therapy supports this hypothesis [14]. Thus, the aim of this study was to verify the associations between the MTHFR C677T polymorphism genotype and the DNA methylation of three candidates genes (COX-2, CDKN2A, and HMLH1), largely associated with gastric carcinogenesis, and the influence of H. pylori strains in this process.
Materials and methods
Clinical specimens
The present study was approved by the Hospital Ethics Committee in the Federal University of Ceará and all subjects signed the informed consent form before inclusion. Samples from 71 patients with gastric adenocarcinoma who had undergone gastrectomy were collected from Walter Cantídio University Hospital and Santa Casa de Misericórdia Hospital, both located in Fortaleza, the Ceará state capital. The histological classification was done according to Lauren's classification by our team of pathologists.
DNA extraction
Genomic DNA was extracted from frozen tumor tissue using the cetyltrimethyl ammonium bromide (CTAB) technique, adapted from Foster and Twell [15]. The DNA extraction was done only in fragments that showed more than 80% of tumor cells. The DNA quality was analyzed by 1% agarose gel electrophoresis and the amount was determined using the NanoDropTM® 3300 fluorospectrometer (Wilmington, DE, USA).
Sodium bisulfite treatment and methylation-specific PCR (MS-PCR)
Extracted DNA of tumor tissue was modified by sodium bisulfite to determine the methylation status of the CDKN2A, HMLH1, and COX-2 genes by MS-PCR, as previously described by Ferrasi et al. [16]. The primers targeted to the studied promoter gene regions are described in Table 1. PCR was performed in 25 μL reaction volume, containing 1× Platinum Taq buffer, 3.0 mM MgCl2 (CDKN2A) or 1.5 mM MgCl2 (HMLH1 and COX-2), 0.4 mM of each dNTPs, 0.64 μM (CDKN2A), 0.24 mM (HMLH1) or 0.4 mM (COX-2) of each primer set, 1U of Platinum Taq DNA Polymerase® (Invitrogen, Foster, CA, USA), and 1 μL of treated DNA. DNA methylated in vitro by Sss-I methylase® (New England Biolabs, Beverly, MA, USA) was used as a positive control. Water and DNA from peripheral lymphocytes of healthy donors were used as negative controls. The PCR products were separated in 6% non-denaturing polyacrylamide gel and subsequently submitted to silver staining (Fig. 1).
For confirmation of the reaction specificity, MS-PCR products from CDKN2A, HMLH1, and COX-2 genes analyzed were cloned with a TOPO TA Cloning Kit® (Invitrogen, California, USA) and both the methylated and unmethylated PCR products were sequenced using an ABI PRISM® BigDye Terminator v.3.0 Cycle Sequencing Kit® (Applied Biosystems, Foster, CA, USA) and ABI Prism 3100 DNA Sequencer® (Applied Biosystems).
Determination of MTHFR C677T genotypes
MTHFR C677T genetic polymorphism was detected by PCR-RFLP as previously described by Frosst et al. [17]. The PCR products of 198 bp were digested with restriction endonuclease Hinf I, producing a 175 and a 23 bp fragments if the polymorphism was present. The digestive products were resolved by electrophoresis in 6% polyacrylamide gel with silver staining. Random samples were reanalyzed for laboratory procedures control (Fig. 2).

Representative electrophoresis of the CDKN2A MS-PCR in 6% polyacrylamide gel with silver staining

Representative electrophoresis of the MTHFR C677T polymorphism in 6% polyacrylamide gel with silver staining

Representative electrophoresis of the ureC and cagA genes amplification in 6% polyacrylamide gel with silver staining
Detection of H. pylori and vacA alleles and the presence of cagA gene
H. pylori infection was detected by PCR of the ureC gene. The primers for the H. pylori studied genes and their annealing temperature are described in Table 2. For confirmation of the reaction specificity, PCR products from ureC gene were cloned with TOPO TA Cloning Kit® (Invitrogen, California, USA) and sequenced using the ABI PRISM® BigDye Terminator v.3.0 Cycle Sequencing Kit® (Applied Biosystems, Foster, CA, USA) and ABI Prism 3100 DNA Sequencer® (Applied Biosystems).
The ureC, cagA, vacA s1/s2, vacA m1/m2 genes were amplified in a 25-μL volume containing 2.5 μL of 10× PCR buffer® (Invitrogen, Cergy Pontoise, France); 1% Tween 20, 1.5 mM of MgCl2 (Invitrogen), 200 μM (each) of dNTPs (Invitrogen), 1U of Platinum Taq polymerase® (Invitrogen); 0.4 μM (ureC, cagA, vacA s1/s2, vacA m1), 0.3 μM (vacA m2) for each primer and 1 μL of DNA. DNAse-free water was used as negative control. The PCR products were analyzed by 1% agarose gel electrophoresis with ethidium bromide staining (Fig. 3). vacA and cagA genes were considered positive when a specific fragment was detected (Table 2). Random samples were reanalyzed to confirm the results. DNA preservation has been confirmed by amplification of different genes in other approaches under study in our laboratory.
Statistical analyses
These were carried out using the statistical program SPSS® version 15.0 (Chicago, IL, USA). Statistically significant differences were evaluated by the chi-square test (χ 2). Correlations were analyzed by Spearman's rank correlation coefficient. The results were considered statistically significant when p values were less than 0.05.
Results
Study population
Among the 71 cases analyzed, 45 (63.4%) were males and 26 (36.6%) females. The average age was 64.2, ranging from 23 to 92 years old. The most frequent site was the gastric non cardia [52/71 (73.2%) × 19/71 (26.8%)]. Intestinal and diffuse subtypes presented 43 (60.5%) and 28 (39.5%) frequencies, respectively.
Frequency of promoter methylation of three CpG islands and MTHFR genotypes
All 71 tumor samples were available for MS-PCR analysis and for MTHFR genotyping. CpG island methylation was found in 30 of the tumors samples (42.2%) for CDKN2A, 38 (53.5%) for COX-2 and 22 (30.9%) for HMLH1. The MTHFR C677T genotype shows similar frequency for the homozygous CC (34/71; 47.8%) and heterozygous CT (31/71; 43.6%) carriers. Homozygous carries for MTHFR 677TT was found in only 8.6% of the patients.
Association between MTHFR C677T, promoter methylation and clinicopathological parameters
The association of each MTHFR C677T genotype to the promoter CpG islands methylation status is shown in Table 3. No significant correlation was found between the promoter methylation for all the studied genes, age (cutoff of 60 years old), histological subtypes (intestinal and diffuse), and MTHFR genotype (Table 3).
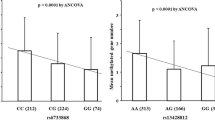
When the tumors were analyzed according to its localization, in the distal tumors a positive correlation was found between the promoter methylation of CDKN2A and the allele T carriers (CT and TT genotypes; r = 0.357, p = 0.009) (Table 4). Considering the patients age with 60 years old or older, this correlation was even higher (r = 0.417, p = 0.014).
Influence of H. pylori genotype in the methylation process
H. pylori infection was present in 68 of the patients (95.8%). Among them, 31 (45.5%) were cagA+/vacAs1m1. As most of the patients were H. pylori positive, we verify if there was an association between the H. pylori strains and the MTHFR genotype in gene methylation, considering clinicopathological parameters. From these analysis, we found a high association between the promoter methylation of CDKN2A and the MTHFR 677T carrier (r = 0.484, p = 0.026) in the distal tumors infected by H. pylori cagA+/vacAs1m1.
Association between number of methylated CpG islands and MTHFR genotype
To investigate whether the MTHFR genotype might be associated with methylation of multiple CpG sites we grouped all the 71 subjects according to the numbers of methylated genes: none, 1, 2, and 3. No promoter methylated gene was present in 20 (28.2%) of the cases while the presence of 1, 2, and 3 was observed in 19 (26.8%), 25 (35.2%), and 7 (9.8%) of the cases, respectively. The mean number of methylated CpG sites according to the MTHFR genotype was 1.12 for CC, 1.42 for CT, 1.33 for TT and 1.40 for T carrier. No significant association was found between MTHFR genotype and number of methylated CpG sites and no statistical difference was verified between the number mean of methylated CpG sites over the different MTHFR genotypes.
Discussion
The relationship between the hypermethylation of tumor suppressor genes or DNA repair genes with the gastric carcinogenesis process has been pointed in many studies; however, the factors involved in the induction of this process are still unknown [18, 19]. Some studies indicate that DNA methylation depends on environmental factors such as H. pylori infection and may also vary according to the presence of polymorphisms involved in this process as those of enzymes involved in the folate cycle [8, 12, 13]. However, there are only few studies focusing these aspects and they present controversial results. So that, in the present study, the association between the methylation status of CDKN2A, HMLH1, and COX-2, the MTHFR C677T polymorphism and the influence of high pathogenic H. pylori strains in gastric cancer were analyzed.
The first analysis focused the association between the presence of promoter methylation and the MTHFR C677T polymorphism. Some studies demonstrated that genetic and epigenetic changes in gastric carcinoma differ depending on the tumor localization and on its histological subtype [20, 21]. Based on that, the data were analyzed according to these aspects. Although no difference was observed regarding to histological subtype, differences were identified according to the tumor anatomic site. In the distal tumors, an unexpected positive correlation was found between the CpG island methylation of CDKN2A and the allele T carriers. In fact, this correlation was even higher when we considered the 60 years old or older patients. Tahara et al. [12] also observed a similar correlation between the hypermethylation of CDKN2A and the MTHFR 677TT genotype, but these authors did not associate it to the tumor anatomic site. Both ours and Tahara et al.'s studies disagree with Weng et al. [22], which found a promoter hypermethylation of CDKN2A in patients with the MTHFR 677CC genotype. Various studies have been associating the MTHFR C677T polymorphism to gastric cancer risk in a specific anatomic localization. Wang et al. [23], evaluating the association of MTHFR C677T and thymidylate synthase promoter polymorphisms with genetic susceptibility found a gastric cardia adenocarcinoma risk related to the MTHFR 677TT and 2R2R genotypes. Likewise, Miao et al. [5] observed that subjects with the MTHFR 677TT variant genotype had a twofold increased risk of gastric cardia adenocarcinoma. These studies suggested that the common functionally polymorphism MTHFR C677T plays a substantial influence in the gastric cardia adenocarcinoma carcinogenesis. However, both studies did not evaluate the methylation pattern of specific genes associated to this polymorphism. Related to methylation status associated with tumor region there are some controversy. Vo et al. [24] found no distinct methylation status for CDKN2A regarded to the tumor localization, while recent studies carried out in Turkey [25] and Germany [26] correlate methylation of promoter CDKN2A to cardiac tumors, but the MTHFR C677T polymorphism influence was not assessed. We suggest that in distal tumors, the silencing of tumor suppressor genes by their promoter CpG islands methylation, especially when influenced by MTHFR polymorphism, might be an important carcinogenic pathway.
It has been well established over the literature that, in general, the methylation status of promoter CpG islands tends to increase with age [27]. From that standpoint, we found that the correlation between methylation of CDKN2A and MTHFR 677T carrier subjects in distal tumors is higher in an older generation (60 years old or older). It seems that the MTHFR polymorphism may have a greater influence on the methylation status as time goes by. Additionally, in the present study, MTHFR C677T polymorphism does not act homogeneously over all the studied genes. MTHFR 677TT genotype seems to be related to the hypermethylation of CDKN2A; on the other hand, it does not show an involvement regarding to HMLH1 and COX-2 methylation status. Some studies have been reporting this dual behavior of MTHFR C677T polymorphism regarding to methylation or unmethylation of promoter regions depending on the target gene [8, 12]. Our data suggests that this differential methylation status influenced by MTHFR C677T genotype depends on the tissue, specifically on the tumor localization. Probably the induction of the methylation process of some genes, like HMLH1 and COX-2, is independent on the MTHFR C677T polymorphism.
Although the hypermethylation of promoter regions in genes involved on gastric carcinogenesis has been well accepted and some studies associate it with MTHFR C677T polymorphism, the influence of H. pylori in this process is still not understood. In the present study, the infection by a highly pathogenic H. pylori strain (cagA + and vacA s1m1 genotype) was associated to the methylation of CDKN2A in the distal stomach in patients with MTHFR 677TT genotype.
Recently, there have been reports showing that H. pylori infection is the most predisposing factor for gene methylation in the stomach [28–30]. It is well known that H. pylori induces chronic inflammation, resulting in down-regulation of the some genes that maintain gastric mucosal homeostasis [14]. Also, the inflammation induced by H. pylori infection seems to promoter methylation of certain genes [31], which could justify an increased risk of tumor formation, mainly associated with the risk of noncardia gastric cancer.
In conclusion, the DNA methylation in promoter regions of CDKN2A associated to the MTHFR 677T carrier is suggested to be a distal tumor characteristic, especially in those 60 years old or older, and depends on the infection by H. pylori cagA/vacAs1m1 strains. More studies with large sample size involving the folate enzymes polymorphisms, DNA methylation and H. pylori infection are needed to extend our understanding of gastric carcinogenesis.
References
Lao-Sirieix P, Caldas C, Fitzgerald RC (2010) Genetic predisposition to gastro-oesophageal cancer. Curr Opin Genet Dev 20(3):210–217
Ministério da Saúde do Brasil. Instituto Nacional de Câncer (2009) Estimativa 2010: incidência de câncer no Brasil. INCA, Rio de Janeiro
Cotran RS et al (2000) Robbins—patologia estrutural e funcional. Guanabara Koogan, Rio de Janeiro
Choi SW, Mason JB (2000) Folate and carcinogenesis: an integrated scheme. J Nutr 130:129–132
Miao X, Xing D, Tan W, Qi J, Lu W, Lin D (2002) Susceptibility to gastric cardia adenocarcinoma and genetic polymorphism in methylenetetrahydrofolate reductase in an at-risk Chinese population. Cancer Epidemiol Biomark Prev 11(11):1454–1458
Götze T, Röcken C, Röhl FW et al (2007) Gene polymorphisms of folate metabolizing enzymes and the risk of gastric cancer. Cancer Lett 251(2):228–236
Shen H, Xu Y, Zheng Y et al (2001) Polymorphisms of 5, 10-methylenetetrahydrofolate reductase and risk of gastric cancer in a Chinese population: a case-control study. Int J Cancer 5:332–336
Wang J, Sasco AJ, Fu C et al (2008) Aberrant DNA methylation of P16, MGMT, and hMLH1 genes in combination with MTHFR C677T genetic polymorphism in esophageal squamous cell carcinoma. Cancer Epidemiol Biomark Prev 17(1):118–125
Graziano F, Kawakami K, Ruzzo A et al (2006) Methylenetetrahydrofolate reductase 677C/T gene polymorphism, gastric cancer susceptibility and genomic DNA hypomethylation in an at-risk Italian population. Int J Cancer 118(3):628–632
Boccia S, Hung R, Ricciardi G et al (2008) Meta- and pooled analyses of the methylenetetrahydrofolate reductase C677T and A1298C polymorphisms and gastric cancer risk: a huge-GSEC review. Am J Epidemiol 167(5):505–516
Perri F, Cotugno R, Piepoli A et al (2007) Aberrant DNA methylation in non-neoplastic gastric mucosa of H. pylori infected patients and effect of eradication. Am J Gastroenterol 102(7):1361–1371
Tahara T, Shibata T, Nakamura M et al (2009) MTHFR 677T carrier influences the methylation status of H. pylori-infected gastric mucosa in older subjects. Dig Dis Sci 54(11):2391–2398
Atherton JC (1998) H. pylori virulence factors. Br Med Bull 54(1):105–120
Atherton JC (2006) The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu Rev Pathol 1:63–96
Foster GD, Twell DJ (1996) Plant gene isolation: principles and practice. Wiley, England
Ferrasi AC, Pinheiro NA, Rabenhorst SH et al (2010) Helicobacter pylori and EBV in gastric carcinomas: methylation status and microsatellite instability. World J Gastroenterol 16(3):312–319
Frosst P, Blom HJ, Milos R et al (1995) A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet 10:111–113
Kim H, Kim YH, Kim SE, Kim NG, Noh SH, Kim H (2003) Concerted promoter hypermethylation of hMLH1, p16INK4A, and E-cadherin in gastric carcinomas with microsatellite instability. J Pathol 200(1):23–31
Shim YH, Kang GH, Ro JY (2000) Correlation of p16 hypermethylation with p16 protein loss in sporadic gastric carcinomas. Lab Invest 80(5):689–695
Driessen A, Nafteux P, Lerut T et al (2004) Identical cytokeratin expression pattern CK7+/CK20− in esophageal and cardiac cancer: etiopathological and clinical implications. Mod Pathol 17(1):49–55
Tahara E (2004) Genetic pathways of two types of gastric cancer. IARC Sci Publ 157:327–349
Weng YR, Sun DF, Fang JY, Gu WQ, Zhu HY (2006) Folate levels in mucosal tissue but not methylenetetrahydrofolate reductase polymorphisms are associated with gastric carcinogenesis. World J Gastroenterol 12(47):7591–7597
Wang LD, Guo RF, Fan ZM et al (2005) Association of methylenetetrahydrofolate reductase and thymidylate synthase promoter polymorphisms with genetic susceptibility to esophageal and cardia cancer in a Chinese high-risk population. Dis Esophagus 18(3):177–184
Vo QN, Geradts J, Boudreau DA, Bravo JC, Schneider BG (2002) CDKN2A promoter methylation in gastric adenocarcinomas: clinical variables. Hum Pathol 33(12):1200–1204
Ksiaa F, Ziadi S, Amara K, Korbi S, Trimeche M (2009) Biological significance of promoter hypermethylation of tumor-related genes in patients with gastric carcinoma. Clin Chim Acta 404(2):128–133
Sarbia M, Geddert H, Klump B, Kiel S, Iskender E, Gabbert HE (2004) Hypermethylation of tumor suppressor genes (p16INK4A, p14ARF and APC) in adenocarcinomas of the upper gastrointestinal tract. Int J Cancer 111(2):224–228
van Rijnsoever M, Grieu F, Elsaleh H, Joseph D, Iacopetta B (2002) Characterisation of colorectal cancers showing hypermethylation at multiple CpG islands. Gut 51(6):797–802
Nardone G, Compare D, De Colibus P, de Nucci G, Rocco A (2007) Helicobacter pylori and epigenetic mechanisms underlying gastric carcinogenesis. Dig Dis 25(3):225–229
Kitajima Y, Ohtaka K, Mitsuno M et al (2008) Helicobacter pylori infection is an independent risk factor for Runx3 methylation in gastric cancer. Oncol Rep 19(1):197–202
Maekita T, Nakazawa K, Mihara M et al (2006) High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res 12(3 Pt 1):989–995
Katayama Y, Takahashi M, Kuwayama H (2009) Helicobacter pylori causes runx3 gene methylation and its loss of expression in gastric epithelial cells, which is mediated by nitric oxide produced by macrophages. Biochem Biophys Res Commun 388(3):496–500
Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci 93:9821–9826
Kang GH, Shim YH, Ro JY (1999) Correlation of methylation of the HMLH1 promoter with lack of expression of HMLH1 in sporadic gastric carcinomas with replication error. Lab Invest 79(7):903–909
Akhtar M, Cheng Y, Magno RM et al (2001) Promoter methylation regulates Helicobacter pylori-stimulated cyclooxygenase-2 expression in gastric epithelial cells. Cancer Res 61(6):2399–2403
Lage AP, Godfroid E, Fauconnier A et al (1995) Diagnosis of Helicobacter pylori infection by PCR: comparison with other invasive techniques and detection of cagA gene in gastric biopsy specimens. J Clin Microbiol 33:2752–2756
Atherton JC, Cao P, Peek RMJR, Tummuru MK, Blaser MJ, Cover TL (1995) Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem 270:17771–17777
Domingo D, Alarcon T, Pietro N, Sanchez I, Lopez-Brea M (1999) cagA and vacA status of Spanish Helicobacter pylori clinical isolates. J Clin Microbiol 37:2113–2114
Conflict of interest statement
We attest the inexistence of conflicts of interest, corporate involvement, or patent holdings.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Neves Filho, E.H.C., Alves, M.K.S., Lima, V.P. et al. MTHFR C677T polymorphism and differential methylation status in gastric cancer: an association with Helicobacter pylori infection. Virchows Arch 457, 627–633 (2010). https://doi.org/10.1007/s00428-010-0996-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-010-0996-3