
Abstract
We present four patients with vasculitis manifesting with unusual clinical or pathological features, generating surgical problems. Two cases presented with pulmonary hypertension, with investigations and radiological evidence prompting clinical suspicion of pulmonary thrombo-embolic disease. First case, with an antecedant history of Wegener's granulomatosis (WG), demonstrated following “embolectomy”, WG involving the large pulmonary elastic arteries. The second case of inoperable “pulmonary thrombo-embolic disease” was subsequently found at limited post mortem to have giant cell arteritis, which affected widespread small peripheral pulmonary arterial vessels. The other two cases were of aortitis occurring in the background of immune-mediated disease, which had been treated with aggressive immunosuppression regimens. The first of these was a case of Cogan's syndrome complicated by descending aortitis, a rarely reported phenomenon, with co-existent acute endocarditis of the aortic valve leaflets. Most cases of endocarditis in this context occur secondary to and in continuity with ascending aortitis. That this case, and a case of ascending aortitis occurring in the context of relapsing polychondritis occurred in the face of aggressive immunosuppression with an apparent clinical response, underscores the need to not accept a clinical picture at face value. This has implications for clinical management, particularly in the follow-up of surgical prosthetic devices such as grafts which may be used in these cases. All four cases emphasise the continued importance of histology and the post-mortem examination in elucidating previously undetected or unsuspected disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Vasculitis both as an isolated phenomenon, or as part of a syndrome affecting multiple tissue types, has protean manifestations and may affect any size vessel. Vasculitis affecting large vessels is notable for its ability to cause significant vascular compromise. Vasculitic disease is also known to mimic other forms of inflammatory disease; a diagnostic problem that can be compounded when well-characterised disease entities present in a nonstereotypic fashion. Additionally, less common forms of vasculitis resulting from autoimmunity, like that in Cogan's syndrome (CS), remain unusual entities where the full spectrum of disease is still being explored. In this context, the unusual individual case of vasculitis may yet inform and expand our knowledge of disease. Immunosuppression is frequently utilised to retard the inappropriate inflammatory activity, with the effectiveness of therapy being quantified by clinical response and monitoring of serological markers of inflammation. In many autoimmune diseases where vasculitis is a possibility, the cardiovascular system is not routinely monitored and involvement can be missed.
With this backdrop, we present four cases of thoracic vasculitis presenting with or displaying unusual clinical features, each eventually coming to surgery and/or autopsy with material subsequently available for histologic examination. The pivotal role of histology and the autopsy is highlighted, illustrating its ability to reveal previously unsuspected disease. Two cases presented with pulmonary hypertension, prompting strong clinical suspicion of pulmonary thrombo-embolic disease with subsequent surgical intervention to remove the “emboli”. The other two cases are those of an aortitis occurring in the background of an immune-mediated disease process ultimately requiring surgery in spite of aggressive immunosuppression.
Materials and results
Case 1
A 34-year-old female, with a known diagnosis of Wegener's granulomatosis (WG), presented in early 2001 with a history of increasing dyspnoea without chest pain or haemoptysis. A histologically proven diagnosis of WG was made in 1993 with an initial presentation of persistent nasal obstruction. Antineutrophil cytoplasmic antibody, antiproteinase 3 had been documented as positive on multiple occasions and her therapy included intravenous cyclophosphamide, azathioprine, corticosteroids and various antibiotics. Echocardiogram revealed an elevated pulmonary artery pressure estimated at 90 mmHg with right ventricular and atrial dilatation and tricuspid regurgitation. Computed tomography pulmonary angiography showed smooth soft tissue filling defects within the right and left main pulmonary arteries (MPA) with extension from the origin of the right MPA into the upper and lower lobe branches. In the left MPA, only focal severe stenosis was seen. These features were interpreted as evidence of thrombo-embolism and a surgical pulmonary “embolectomy” was performed. At surgery, hard adherent tissue was seen within the proximal pulmonary artery and this was submitted for histopathology.
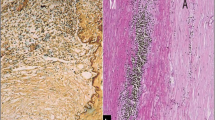
Microscopically, the specimen showed vasculitis of the pulmonary artery (see Fig. 1). There was a mixed inflammatory cell infiltrate with abundant lymphocytes, plasma cells and occasionally foamy histiocytes within the artery wall (see Fig. 2) with areas of basophilic geographic necrosis surrounded by palisading histiocytes and multinucleate giant cells. Stains for mycobacteria and fungi were negative. The appearances were of a giant cell vasculitis with features in keeping with Wegener's granulomatosis affecting the main pulmonary artery.

Muscularized small pulmonary artery from Case 1 showing intimal proliferation and destruction of the elastic lamina with replacement by fibrosis. Elastic von Gieson ×5

High power photomicrograph of the same artery from Case 1 in Fig. 1 showing histiocytes and giant cells with accompanying mononuclear inflammatory infiltrate HE ×20
Case 2
A 45-year-old female with a history of Crohn's disease and systemic hypertension presented in 1995 with shortness of breath and was transferred to the Royal Brompton Hospital, London, with a clinical diagnosis of thrombo-embolic pulmonary hypertension. An angiogram performed on admission revealed occlusion of the right upper lobe, right middle lobe, left upper lobe and left lingula pulmonary arteries. She was treated with insertion of a Greenfield inferior vena cava filter and anticoagulated using Warfarin. Her lupus anticoagulant and anticardiolipin antibodies were strongly positive. 15 months later, she represented with worsening shortness of breath and marked exercise intolerance. Although surgical intervention was attempted, the thrombi occluding the arteries were too peripherally lodged to permit removal. She died shortly after surgery in right-sided heart failure. Post-mortem examination revealed that the left and right main pulmonary arteries showed slightly raised atherosclerotic plaques. The lobar branches were normal. All segmental arteries showed focal narrowing and abrupt occlusion of branches by thrombi and occlusion of peripheral vessels by firm white fibrotic tissue with subpleural scarring. Histology of the segmental arterial branches showed a giant cell vasculitis with destruction of the media complicated by thrombosis with luminal occlusion. Smaller muscular vessels throughout the lung parenchyma showed luminal occlusion by fibrous tissue with evidence of recanalisation and replacement of vessel wall by fibrosis. There was no evidence of granulomas in the lung parenchyma, heart or lymph node and no evidence of generalised vasculitis in the coronary vessels, the aorta and its branches. The features were those of a giant cell arteritis with associated thrombosis. Because the autopsy was limited to the chest, it was not possible to assess if there was vasculitis affecting other organs.
Case 3
A 38-year-old female with a diagnosis of Cogan's syndrome made 3 months earlier developed palpitations. Echocardiogram showed good left ventricle function with mild to moderate aortic regurgitation and mild mitral regurgitation. An initially normal cardiovascular exam was documented. Blood tests showed raised ESR and CRP, an autoimmune serology screen was negative. Abdominal imaging revealed an aneurysmal dilation of the descending aorta with a maximal measurement of 4 cm. in the antero-posterior dimension. Immunosuppression was increased and included steroids and cyclophosphamide. One month later, she was admitted to hospital with biventricular failure and severe acute aortic regurgitation and an immediate salvage aortic valve replacement was performed. One cusp was sent for microbiological culture and failed to grow an organism. At histology, the other two cusps grossly showed tiny cream-coloured foci on the surface. Microscopically, there was severe acute endocarditis with microabscess formation and fibrinoid necrosis (see Fig. 3). Special stains for organisms were negative. She died shortly after surgery from an intracerebral haemorrhage. At post-mortem examination, the ascending aorta and the great vessels of the head and neck were normal. Distal to the origin of the left subclavian artery the thoracic aorta was dilated with a circumference of 70 mm. The intima showed a “cobble-stoned” appearance and the wall was thickened (see Fig. 4). There was no ulceration or thrombosis in the aorta. Microscopic examination of the ascending aorta and aortic arch vessels was normal. The descending aorta distal to the subclavian artery origin showed marked aortitis with intimal proliferation and fibrosis, as well as infiltration of the intima and adjacent media by neutrophil leukocytes surrounding areas of medial necrosis with focal transmural destruction in the upper abdominal aorta. The vasa vasorum extended into the media and were surrounded by a chronic inflammatory, predominantly lymphocytic, infiltrate. Macrophages were prominent, with very occasional giant cells noted. These changes extended from the descending thoracic aorta into the upper and lower abdominal aorta and involved the mesenteric vessels and the iliac vessels, with sparing of the renal vessels. The brain was not examined at the family request. The features were those of a florid acute on chronic aortitis with aortic valve endocarditis complicating Cogan's syndrome.

Aortic valve cusp from Case 3 showing extensive acute inflammation HE ×4

Aorta from Case 3 showing marked wall thickening with intima showing a cobblestoned appearance
Case 4
A 54-year-old woman was diagnosed with relapsing polychondritis with involvement of ear and nasal cartilage as well as scleritis. An echocardiogram revealed mild aortic regurgitation. In 1999, she experienced a flare up of her disease, with scleritis, worsened aortic regurgitation and elevation of serological markers of inflammatory activity. She was managed with increased immunosuppression with steroids and azathioprine. Subsequent echocardiogram revealed worsening aortic regurgitation with dilation of the aortic root and an ascending aortic aneurysm, prompting the introduction of cyclophosphamide to her immunosuppressant regime. Her ESR was decreased from 90 to 24 mm/h over 4 months, and her immunosuppressant dose was reduced. At this stage magnetic resonance imaging of the chest showed an aneurysm of the ascending aorta extending from the sinotubular junction to beyond the right innominate artery. One month later, aortic valve replacement and aortic root replacement were performed. Pathological examination of the aorta showed a thickened wall measuring 5 mm with pronounced irregularity of the intimal surface (see Fig. 5). Histologically, intimal fibrosis and infiltration by acute and chronic inflammatory cells was noted. The medial elastic tissue was destroyed and replaced by vascular granulation tissue and focal necrosis. The vasa vasorum extended throughout the aortic wall, with surrounding chronic inflammation and endothelial swelling. Neutrophil leukocyte aggregates forming microabscesses were present in the media and intima. The adventitia showed focal lymphoid infiltration and fibrosis. Special stains for bacteria and fungi were negative. No giant cells, granulomas or eosinophils were present. The features were consistent with an aortitis associated with polychondritis.

Aorta from Case 4 showing marked intimal thickening and wall fibrosis
Discussion
Vasculitic disorders can present in many ways as highlighted by these thoracic cases and recognition of these diverse manifestations is important. Vasculitides are classified according to the size of the involved vessels. The pathological patterns of large vessel vasculitides are Buerger's disease, temporal arteritis, Takayasu's disease, Behcet's disease, infectious arteritides and rheumatologic diseases [1]. Buerger's disease is a thrombotic arteriopathy with no arterial wall involvement. Temporal arteritis and Takayasu's disease belong to the group of giant cell arteritides. In temporal arteritis, the inflammation is prominent in the internal part of the media with intimal thickening and subsequent occlusion. In Takayasu's disease, the external part of the media is prominently involved with fibrous thickening of the arterial wall with stenosis. Rheumatologic diseases usually result in giant cell aortitis with aortic valve incompetence. The first two cases illustrate that vasculitis involving the pulmonary arteries can mimic thrombo-embolism with progression to surgery in both cases. Vasculitis of the pulmonary artery has been attributed to either Takayasu's arteritis [2, 3], or giant cell arteritis [4], with patients presenting with evidence of pulmonary artery obstruction mimicking thrombo-embolic disease, pulmonary hypertension and severe haemoptysis. WG does not usually involve the large elastic arteries and to our knowledge this is the first reported case of WG affecting the main pulmonary arteries, here presenting clinically as a mimic of pulmonary thrombo-embolic disease. There has been one case report of a link between Wegener's granulomatosis and Takayasu's aortitis found in an autopsy case [5] and an overlap with giant cell arteritis has been reported in atypical presentations of Wegener's granulomatosis [6]. In case 2, the history of Crohn's disease may be important since this is linked to granulomatous lesions in airways and elsewhere [7] but again there has been no previous link to pulmonary artery vasculitis. As in the WG case, there has one case report linking Crohn's disease to aortitis [8]. The finding of antiphospholipid antibody pointed to thrombosis with microvascular angiopathy in the lung but there is no link to giant cell vasculitis. There has been one previous report of similar giant cell vasculitis involving the pulmonary artery and aorta in a young woman presenting with signs and symptoms of chronic thrombo-embolic pulmonary hypertension who also underwent pulmonary thrombo-endarterectomy with concomitant coronary artery bypass [9]. It is important to differentiate forms of vasculitis from chronic thrombo-embolic pulmonary hypertension because management of these differ considerably. The outcome for both the above patients could have been different if vasculitis had been considered.
Granulomatous vasculitis due to infection is always an important diagnosis to exclude, particularly in patients treated with immunosuppressive agents.
The two cases of aortitis emphasise that involvement of large vessels occurs in these rare autoimmune diseases of Cogan's syndrome and relapsing polychondritis, and that vasculitis can progress despite diagnosis and therapy, requiring surgery in both cases. These two cases also emphasise that both acute and chronic inflammatory cells are seen in blood vessels in these conditions.
Typical CS is a disease of young adults consisting of flares of interstitial keratitis and vestibuloauditory dysfunction with deafness. The demonstration of autoantibodies to inner ear and endothelial antigens [10] indicates that autoimmunity could initiate or perpetuate an abnormal immune response. The prognosis of typical CS is excellent but life-threatening aortic insufficiency develops in 10% of reported cases [11]. The features of our case of CS are noteworthy for several reasons. The acute aortic valve endocarditis occurred in the background of intensive immunosuppressive therapy, making the exclusion of infective aetiology mandatory. Also, despite therapy, there was florid acute and chronic inflammation in both the aortic valve leaflets and the aortic wall. Most valve lesions mentioned in case reports of CS, occur with a contiguous ascending aortitis, a feature not present in this case. Involvement of the aortic cusps, with an uninvolved ascending aorta and an acute descending and abdominal aortitis has not been described before.
Relapsing polychondritis (RP) is a rare multisystem autoimmune disease of unknown origin characterised by recurrent episodes of inflammation and progressive destruction of cartilaginous tissues. Additionally proteoglycan-rich structures of the eye, heart, blood vessels or inner ear may all be affected. In most patients, RP manifests in a fluctuating but progressive course. An immunological basis is hypothesised following the demonstration of serum antibodies to type II collagen in up to two thirds of cases. Cardiovascular complications are documented in 25% of patients and include aortic and mitral regurgitation, aortic aneurysm, aortic dissection, myocarditis, pericarditis, atrioventricular block and systemic vasculitis [12]. The aortitis is usually lymphocytic infiltration around the vasa vasorum of the media accompanied by fragmentation and loss of the elastic tissue with fibrous replacement. Of special interest in this case is the presence of florid acute inflammation of all three layers of the aortic wall with microabscess formation despite aggressive immunosuppression with clinical improvement and a documented decline in serological markers of inflammatory activity, highlighting the importance of careful cardiological follow up in these cases. Therefore, we recommend close and prolonged follow-up: firstly because there can be early paravalvular prosthetic leakage due to the friability of the tissue to which it has been anchored in all cases of aortitis; secondly because aortic aneurysms occur frequently in relapsing polychondritis, may be multiple, may involve all parts of the aorta and result in fatal rupture even in asymptomatic patients; and thirdly because there can be a fatal outcome due to other organ involvement, like airway obstruction, acute glomerulonephritis or systemic vasculitis.
In conclusion, this paper reports four unusual cardiopulmonary vascular complications of four different diseases, all with an autoimmune basis, leading to surgery in each case. Two of these cases presented clinically and radiologically with evidence of pulmonary thrombo-embolic disease. One of the cases, with an antecedent history of Wegener's granulomatosis, was shown following “embolectomy”, to have a WG lesion affecting the large pulmonary elastic arteries, a finding heretofore not reported in the literature. The second case of “pulmonary thrombo-embolic disease” was found at limited post mortem to have giant cell arteritis affecting widespread small peripheral pulmonary arterial vessels. The association with positive serology for anticardiolipin antibodies, a finding suggestive of primary antiphospholipid syndrome, has also previously not been reported. We further reported a case of Cogan's syndrome, complicated by descending aortitis, a rarely reported phenomenon, with co-existent acute endocarditis of the aortic valve leaflets. This scenario, too, has not been previously reported, with the occasional cases with aortic valve inflammation usually arising consequent to and in continuity with ascending aortitis. That this case and the ascending aortitis occurring in relapsing polychondritis described above occurred in the face of an aggressive immunosuppressive regimen including cyclophosphamide with an apparent clinical response, again underscores the need to not accept a clinical picture at face value. A similar point can be made with the WG case where the patient developed pulmonary vasculitis despite therapy. If patients develop new symptoms related to the cardiovascular system they should be clinically investigated. These cases emphasise the need for the surgeon to be aware of these rare entities and plan his surgery accordingly and also emphasise the importance of histology and the post-mortem examination in elucidating previously undetected or unsuspected disease.
References
Bruneval P (1999) Pathology of large vessel vasculitides. Rev Med Interne 20:875–887
Koyabu S, Isaka N, Yada T et al (1993) Severe respiratory failure caused by recurrent pulmonary hemorrhage in Takayasu's arteritis. Chest 104:1905–1906
Elsasser S, Soler M, Bolliger C et al (2000) Takayasu disease with predominant pulmonary involvement. Respiration 67:213–215
Chassagne P, Gligorov J, Dominique S (1995) Pulmonary artery obstruction and giant cell arteritis. Ann Intern Med 122:732
Mejia-Hernandez C, Alvarez-Mendoza A, DeLeon-Bojorge B (1999) Takayasu's arteritis coexisting with Wegener's granulomatosis in a teenager with renal insufficiency: case report. Pediatr Dev Pathol 2:385–388
Leavitt RY, Fauci AS (1992) Less common manifestations and presentations of Wegener's granulomatosis. Curr Opin Rheumatol 4:16–22
Rogler G, Scholmerich J (2004) Extraintestinal manifestations of inflammatory bowel disease. Med Klin (Munich) 99:123–130
Wackerlin A, Zund G, Maggiorini M et al (1997) Aortic valve insufficiency in Crohn disease. Schweiz Med Wochenschr 127:935–939
Brister SJ, Wilson-Yang K, Lobo FV et al (2002) Pulmonary thromboendarterectomy in a patient with giant cell arteritis. Ann Thorac Surg 73:1977–1979
Lunardi C, Bason C, Leandri M et al (2002) Autoantibodies to inner ear and endothelial antigens in Cogan's syndrome. Lancet 360:915–921
Haynes BF, Kaiser-Kupfer MI, Mason P et al (1980) Cogan syndrome: studies in thirteen patients, long-term follow-up, and a review of the literature. Medicine (Baltimore) 59:426–441
Del Rosso A, Petix NR, Pratesi M et al (1997) Cardiovascular involvement in relapsing polychondritis. Semin Arthritis Rheum 26:840–844
Conflicts of interest statement
We declare that we have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jansen, M., Saleh, S., Bolster, M. et al. Thoracic vasculitis presenting as surgical problems. Virchows Arch 456, 91–96 (2010). https://doi.org/10.1007/s00428-009-0865-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-009-0865-0