
Abstract
Plasma protein fibrinogen variants cause fibrinogen Aα-chain (AFib) amyloidosis, which presents with hypertension, proteinuria, and azotemia. Six AFib mutations have been reported thus far. We identified three patients who presented with marked proteinuria and serum creatinine elevations. Their kidney biopsies revealed destruction of the glomerular architecture by amyloid deposits with typical, apple-green birefringence in polarized light after Congo red staining. We found immunoreactivity against fibrinogen, which is typical for this type of amyloidosis. We sequenced the FGA exon 5 and demonstrated heterozygosity for the p.Glu526Val mutation in all three cases. This amino acid substitution is the most common fibrinogen Aα-chain variant causing AFib amyloidosis. The mutation has been reported in individuals of European and American descent but not yet in German patients. AFib amyloidosis should therefore be considered an important differential diagnosis in German patients with renal amyloidosis. In the cases described here, the use of antibodies directed against fibrinogen, followed by direct gene sequencing, revealed the underlying cause.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hereditary amyloidosis is an autosomal-dominant disease characterized by insoluble protein deposits in various organs. Hereditary amyloidosis has been associated with variant apolipoprotein AI, apolipoprotein AII, gelsolin, lysozyme, fibrinogen Aα-chain, or transthyretin proteins. The most frequent form is the transthyretin-derived ATTR amyloidosis, which clinically presents with polyneuropathy and/or cardiomyopathy [3]. These symptoms clearly differ from hereditary renal amyloidosis (HRA), where nephropathy is the main manifestation. Several HRA kindred have been reported since Ostertag [18] first described a German family in 1932. Common HRA proteins are fibrinogen (AFib amyloidosis), lysozyme (ALys amyloidosis), apolipoprotein AI (AApoAI amyloidosis), and apolipoprotein AII (AApoAII amyloidosis). One ApoAI variant was reported to also cause neuropathy [17, 18]. Six mutations in the fibrinogen Aα-chain (FGA) cause AFib amyloidosis. The most common variant is p.Glu526Val, which was first described in 1994 in two Irish families [20]. The sole clinical manifestation in those patients was a nephropathy appearing at age 40–60 years without any sign of neuropathy [21]. Five additional mutations causing AFib amyloidosis were since reported. One was a single base deletion at nucleotide 4897 resulting in p.Thr522fs first described in a French family [10]. A G-to-T transversion leading to the amino acid exchange p.Arg554Leu was observed in a kindred of Peruvian descent [4]. A single base deletion at nucleotide 4904 resulting in p.Ser524fs was found in the US [22]. A p.Glu540Val replacement was described in a German patient [6], and the p.Asp517_Thr522delinsGlnSer mutation was recently found in a 7-year-old Korean girl [13] (Table 1).
Clinically, AFib amyloidosis presents with hypertension, proteinuria, and azotemia. Amyloid is typically found in the renal glomeruli, while the liver and spleen are only involved in more advanced cases [2]. With the exception of one reported case from Portugal, no neuropathy has been observed in AFib amyloidosis. Fibrinogen is involved in the coagulation process; however, the aforementioned variants do not seem to affect the normal function of fibrinogen [19]. Although hereditary amyloidosis is relatively uncommon, 5% of patients referred to the UK National Amyloidosis Center with an apparent diagnosis of acquired AL amyloidosis carried the p.Glu526Val variant of the fibrinogen Aα-chain [14].
Treatment options for this disease are limited. Renal transplantation is associated with rapid recurrence of amyloid deposits in the graft, resulting in subsequent renal failure. Combined renal and hepatic transplantation was reportedly successful in two amyloidogenic variants and could be effective in all AFib amyloid cases. In the presence of a normal liver, the variant fibrinogen is eliminated from the serum, and the amyloid deposits can be mobilized from the affected organs [15, 24]. We describe three German patients with fibrinogen Aα-chain amyloidosis due to the p.Glu526Val mutation. Since pathologists will ultimately make the diagnosis of such patients, their thorough knowledge of the genetic basis is highly important for subsequent therapeutic plans.
Clinical history
Patient no. 1
A 62-year-old woman developed myalgia and low-grade fever over a 20-year period. Hypertension developed after 10 years. Proteinuria of >1 g/day and a creatinine concentration of 2.3 mg/dl were observed 18 years after symptom onset. A renal biopsy was then performed. Amyloidosis of unknown cause and subtype was diagnosed. Rheumatological examination ruled out a periodic fever syndrome. The patient was then referred to the Amyloidosis Clinic, University of Heidelberg. She had no macroglossia and no cutaneous bleeding. The heart, liver, and gastrointestinal tract were not affected. However, the patient showed early signs of a sensory polyneuropathy in fingers and feet, which was confirmed by electrophysiological examination. Laboratory tests showed no monoclonal gammopathy. The free light chain test and a bone marrow biopsy were also normal. The family history provided no evidence of a hereditary amyloidosis or hereditary kidney disease.
Patient no. 2
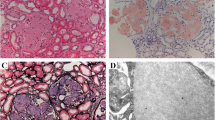
A 49-year-old man without known kidney disease but with hypertension, hyperlipidemia, and coronary artery disease was admitted to hospital with rising serum creatinine levels and severe nephrotic syndrome. On admission, the proteinuria was 15 g/day and the creatinine clearance was 11 ml/min. A kidney biopsy revealed renal amyloidosis (Fig. 1). No other organs were involved. The patient’s mother had an unknown renal disease and died of a myocardial infarction at the age of 61 years.

Kidney biopsy obtained from patient no. 2. Hematoxylin-and-eosin-stained (H & E) sections showed extensive destruction of the glomerular architecture by amorphous eosinophilic material. Congo red staining (CR) and polarization microscopy demonstrated the typical apple-green birefringence of amyloid. The amyloid deposits were immunostained with an antibody directed against fibrinogen (Fib), while no immunostaining was found with antibodies directed against AA amyloid (AA) or λ-light chain (Lambda). Original magnifications 100- (left upper panel) and 200-fold (all other panels). Hematoxylin counterstain
Patient no. 3
A 64-year-old man was admitted to the Amyloidosis Clinic, University of Heidelberg, after a kidney biopsy disclosed amyloidosis. He had developed hypertension 20 years earlier and noted progressive dyspnea for 4 years. His physician observed progressive proteinuria and renal failure that culminated in dialysis. No monoclonal gammopathy, free light chains, or increased plasma cells were found. Amyloidosis elsewhere could not be established in this patient.
Materials and methods
Histology and immunohistochemistry
The kidney biopsies were fixed in formalin and embedded in paraffin. Serial sections were stained with hematoxylin and eosin. Amyloid was detected in Congo-red-stained sections viewed under cross-polarized light. Immunohistochemistry was performed with commercially available monoclonal antibodies directed against AA amyloid (1:600) and polyclonal antibodies against amyloid P-component (1:5,000), fibrinogen (1:2,000), lysozyme (1:3,000), transthyretin (1:4,000), λ-light chain (1:160,000), κ-light chain (1:160,000), and β2-microglobulin (β2 M; 1:2,000; all DAKO, Hamburg, Germany), as well as with noncommercially available polyclonal antibodies directed against apolipoprotein AI (anti-apo-AI; dilution 1:1,000) [9], λ-light chain-derived amyloid proteins (anti-AL1 antibody; dilution 1:3,000) [5], and anti-λ-light chain-peptide antibodies (AL3, dilution 1:250; AL7, 1:500). Immunostaining was done with the BenchMark® XT immunostainer using the ultraView™ Universal Alkaline Phosphatase Red Detection Kit (both Ventana Medical Systems, Inc., Tucson, AZ, USA). Prior to incubation with anti-amyloid P component, -apoAI, -λ and -κ light chain, -transthyretin antibodies, and AL7, sections were pretreated with cell conditioning 1 according to the manufacturer’s instructions (CC1; Ventana). The specificity of immunostaining was verified using specimens containing known classes of amyloid (AA amyloid, apolipoprotein AI, β2 microglobulin, transthyretin, λ-light chain) or by using positive controls recommended by the manufacturers (remaining antibodies). Omission of the primary antibodies served as a negative control.
DNA isolation, PCR amplification, and sequence analysis
The participating subjects gave their written, informed consent prior to the genetic analysis and the ethical committee approved the study. Ethylene diamine tetraacetic acid blood was drawn, and the genomic DNA was isolated from peripheral blood leukocytes using the DNA blood mini kit from Qiagen (Hilden, Germany). Amplification of the 3′ end of FGA exon 5, encoding amino acids 508-611, was performed in a 50-μl volume containing 1× reaction buffer (Qiagen), 1.5 mM MgCl2, 125 μM dNTPs, 400 nM each of the forward and reverse primer, approximately 200 ng genomic DNA, and 2.5 U of HotStartTaq DNA polymerase (Qiagen). The following thermocycling conditions were used: an initial denaturation step of 15 min at 95°C to activate the Taq polymerase, followed by 40 cycles of 95°C for 20 s, 62°C for 20 s, and 72°C for 20 s, and a final extension step of 72°C for 10 min. The polymerase chain reaction (PCR) product size and quantity was analyzed by agarose gel electrophoresis. Fragments were purified with the ExoSAP-IT® kit for PCR product cleanup (USB Corp., Cleveland, OH, USA) and sequenced with the ABI PRISM® BigDye Terminator v3.1 Ready Reaction Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA). Sequences were analyzed on an ABI PRISM® 3130 Genetic Analyzer.
Results
Histology and immunohistochemistry
The kidney biopsies of all three patients showed extensive effacement of the glomerular architecture by amorphous amyloid deposits with typical apple-green birefringence in polarized light after Congo red staining (Fig. 1). The amyloid deposits were purely glomerular in all three patients. The amyloid deposits were immunoreactive for amyloid P component but not for any of the other antibodies directed against AA amyloid, ApoAI, lysozyme, β2-microglobulin, λ- and κ-light chain, or transthyretin. An antibody directed against fibrinogen stained some of the deposits intensely and raised the suspicion of a hereditary AFib amyloidosis in all patients (Fig. 1).
Sequence analysis
All three patients were shown to be heterozygous for a c.1577A>T substitution in exon 5 of the FGA gene, leading to the replacement of glutamic acid (GAG), by valine (GTG) at residue 526. No mutations were found in the genes coding for transthyretin, apolipoprotein AI, apolipoprotein AII, or lysozyme. Furthermore, we established that the patients were not related.
Examination of family members
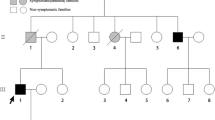
The daughter and son of patient no. 1 are clinically well. Only the daughter underwent mutation screening and harbored the p.Glu526Val mutation (Fig. 2a). The mother of patient no. 2 had an undefined kidney disease and died of myocardial infarction at the age of 61; her DNA was never analyzed. The mother’s sister carries the mutation but is asymptomatic thus far. Her daughter and grandchildren have not undergone genetic testing. Two other siblings died of leukemia and cancer at the age of 50 and 59 years, respectively (Fig. 2b). No information about the death of the patient’s grandparents (generation I) was available. The patient himself has no siblings or children. The father and aunt of patient no. 3 developed chronic kidney disease (Fig. 2c). The patient’s three children are healthy. None of the family members has undergone genetic testing thus far.

Pedigree analysis. a Patient no. 1 is represented by the filled circle in generation I. Her daughter is carrying the p.Glu526Val mutation without any symptoms and her son is healthy and not genetically examined. b Patient no. 2 is represented by the filled square in generation III. The mother suffered from an undefined kidney disease and died of myocardial infarction at the age 61. The mother’s sister carries the p.Glu526Val mutation asymptomatically. It is unknown whether her daughter (generation II) and granddaughters (generation IV) carry the mutation. The other two siblings of the mother (generation II) were not genetically examined and died of leukemia and cancer at the age of 50 and 59 years, respectively. c Patient no. 3 is represented by the filled square in generation III. The patient’s father and aunt suffered from chronic kidney disease but were not genetically examined. Individuals that are not genetically examined are represented by gray squares or circles
Discussion
The pathologist serves as consultant to the clinician. Modern genetic analysis now enables the pathologist not only to make highly specific, mechanistic diagnoses, but also to provide clinically relevant information for the entire family. Furthermore, steady progress is being made in the development of novel amyloidosis therapies. These treatments may also be gene specific in terms of efficacy. The mutations we describe have hitherto fore not been described in Germany.
Fibrinogen is a coagulation plasma protein synthesized mainly in the liver [16]. The fibrinogen molecule is a 45-nm structure consisting of two sets of disulfide-bridged Aα-, Bβ-, and γ-chains. The Aα chain is the largest of the three with 610 amino acids and a molecular weight of 66 kDa [8]. The FGA gene spans over 7 kb and contains six exons. Alternative splicing results in two isoforms, which vary in the carboxyterminus [16]. Although FGA mutations are rare, their effects are profound, namely dysfibrinogenemia, hypofibrinogenemia, afibrinogenemia, or AFib amyloidosis [16]. All six AFib amyloidosis mutations observed thus far are located in exon 5 and affect one ore more residues at amino acid positions 517–554. In patients carrying the p.Arg554Leu variant, the amyloid fibril subunit protein contains the fibrinogen α-chain residues 500–580. A similar deposited peptide is found in p.Glu526Val carriers. The two frame shifts, p.Ser524fs and p.Thr522fs, both result in a premature stop codon in amino acid residue 548 [10, 22]. These four peptides found in amyloid deposits all share the unchanged amino acid sequence 500–521. Serpell et al. [19] believed that this 21-residue fragment is responsible for the formation of amyloid fibrils, although that could not be the case with the indel mutation, affecting amino acids 517–522.
Similar to other forms of hereditary amyloidosis, the mechanisms underlying fibril formation in Aα-chain amyloidosis are principally unknown. In contrast to ATTR amyloidosis, where both the variant and the wild-type protein are able to form amyloid, only the variants of the fibrinogen Aα-chain are amyloidogenic. In some cases of AFib amyloidosis, only the variant protein was found in the blood [4, 10], while in others, the normal peptide was also found [7, 20, 22, 23]. Furthermore, no clear pattern regarding the same type of mutation has been described.
The phenotypic penetrance of AFib amyloidosis is low and most patients do not have a family history [12]. In our cases, for example, the aunt of patient no. 2 is a carrier of the same mutation but does not have any nephropathy. Her daughter and granddaughters were not examined. Although the patient’s mother could not be genetically examined, she probably carried the mutation since she had an undiagnosed renal disease. Similarly, no family history of renal amyloidosis was present for patient no. 1; however, genetic examination of the daughter revealed that she also carried the p.Glu526Val mutation. Probably one of the patient’s parents harbored the mutation, although clinical manifestations were not present or diagnosed.
Our results emphasize the importance of immunohistochemical classification, combined with genetic analysis, in the diagnostic workup of renal amyloidosis. Immunohistochemistry excluded other causes of renal amyloidosis, particularly the AA and AL types, and raised suspicion of AFib amyloidosis. AFib was indeed confirmed by genetic testing in all our patients. The correct classification of the responsible protein is crucial in the clinical decision to perform organ transplantation. Conceivably, combined kidney and liver transplantation provides the solely effective clinical approach to this disease. Diagnosis of HRA can readily be made, for example, by direct DNA sequencing or single-strand conformation polymorphism analysis. The nucleotide substitution resulting in the p.Glu526Val exchange also creates a Mae III restriction endonuclease recognition site and can therefore also be analyzed by restriction enzyme digestion [2]. In our three cases, the use of polyclonal antibodies raised against fibrinogen was suggestive for this type of amyloidosis. Direct sequencing of FGA exon 5 then revealed the responsible p.Glu526Val mutation. The onset of Aα-chain amyloidosis occurs mostly at the age of 30–50 [4, 20, 24]. One African–American kindred showed a later onset (60 years) [22]. Two affected children aged 7 and 12 years also have been reported (Table 1) [13, 10]. One of the patients described here was 63 years old at the time of diagnosis, a slightly later onset than observed in the other patients carrying the p.Glu526Val mutation.
The p.Glu526Val substitution is the most common fibrinogen Aα-chain defect connected with amyloidosis and has been reported in kindred and individuals of European and American descent; however, this mutation has not been previously observed in German patients. HRA was first described in a German family in an abstract in 1932 and as a full paper in 1950. At that time, the molecular cause could not be established [18]. Since then, the only reported amyloidogenic mutation in the fibrinogen Aα-chain gene found in a single German kindred was the p.Glu540Val variant reported in abstract form by Bybee et al. in 2004 [6]. We here describe three additional German patients with the p.Glu526Val variant also leading to AFib amyloidosis, which may be more prevalent in the German population than previously suspected. AFib amyloidosis should therefore be included in the differential diagnosis of every German patient presenting with renal amyloidosis. The diagnostic workup described here, including immunohistochemical classification of the amyloid protein followed by direct DNA sequencing, is reliable and excludes other causes of amyloidosis.
Abbreviations
- FGA:
-
fibrinogen Aα-chain gene
- HRA:
-
hereditary renal amyloidosis
- TTR:
-
transthyretin
- Fib:
-
fibrinogen Aα-chain
- Lys:
-
lysozyme
- ApoAI:
-
apolipoprotein AI
- ApoAII:
-
apolipoprotein AII
References
Alexander F, Atkins EL (1975) Familial renal amyloidosis. Case reports, literature review and classification. Am J Med 59:121–128
Benson MD (2005) Ostertag revisited: the inherited systemic amyloidoses without neuropathy. Amyloid 12:75–87
Benson MD, Kincaid JC (2007) The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 36:411–423
Benson MD, Liepnieks J, Uemichi T, Wheeler G, Correa R (1993) Hereditary renal amyloidosis associated with a mutant fibrinogen alpha-chain. Nat Genet 3:252–255
Bohne S, Sletten K, Menard R, Buhling F, Vockler S, Wrenger E, Roessner A, Rocken C (2004) Cleavage of AL amyloid proteins and AL amyloid deposits by cathepsins B, K, and L. J Pathol 203:528–537
Bybee, A., Hollenbeck, M., Debusman, E., Gopaul, D., Gilbertson, J., and Lachmann, H. J. (2004) Hereditary renal amyloidosis in a German family associated with fibrinogen Aalhpa chain Glu540Val. In: Xth International Symposium on Amyloid and Amyloidosis, Tours, Loire Valley, France,71.
de Carvalho M, Linke RP, Domingos F, Evangelista T, Ducla-Soares JL, Nathrath WB, zevedo-Coutinho C, Lima R, Saraiva MJ (2004) Mutant fibrinogen A-alpha-chain associated with hereditary renal amyloidosis and peripheral neuropathy. Amyloid 11:200–207
Doolittle RF, Watt KW, Cottrell BA, Strong DD, Riley M (1979) The amino acid sequence of the alpha-chain of human fibrinogen. Nature 280:464–468
Gregorini G, Izzi C, Obici L, Tardanico R, Rocken C, Viola BF, Capistrano M, Donadei S, Biasi L, Scalvini T, Merlini G, Scolari F (2005) Renal apolipoprotein A-I amyloidosis: a rare and usually ignored cause of hereditary tubulointerstitial nephritis. J Am Soc Nephrol 16:3680–3686
Hamidi AL, Liepnieks JJ, Uemichi T, Rebibou JM, Justrabo E, Droz D, Mousson C, Chalopin JM, Benson MD, Delpech M, Grateau G (1997) Renal amyloidosis with a frame shift mutation in fibrinogen a alpha-chain gene producing a novel amyloid protein. Blood 90:4799–4805
Hamidi AL, Fournier V, Billerey C, Justrabo E, Chevet D, Droz D, Pecheux C, Delpech M, Grateau G (1998) Fibrinogen A alpha chain mutation (Arg554 Leu) associated with hereditary renal amyloidosis in a French family. Amyloid 5:279–284
Hawkins PN (2003) Hereditary systemic amyloidosis with renal involvement. J Nephrol 16:443–448
Kang HG, Bybee A, Ha IS, Park MS, Gilbertson JA, Cheong HI, Choi Y, Hawkins PN (2005) Hereditary amyloidosis in early childhood associated with a novel insertion–deletion (indel) in the fibrinogen A alpha chain gene. Kidney Int 68:1994–1998
Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, Pepys MB, Hawkins PN (2002) Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med 346:1786–1791
Mousson C, Heyd B, Justrabo E, Rebibou JM, Tanter Y, Miguet JP, Rifle G (2006) Successful hepatorenal transplantation in hereditary amyloidosis caused by a frame-shift mutation in fibrinogen A alpha-chain gene. Am J Transplant 6:632–635
Neerman-Arbez M (2006) Molecular basis of fibrinogen deficiency. Pathophysiol Haemost Thromb 35:187–198
Nichols WC, Dwulet FE, Liepnieks J, Benson MD (1988) Variant apolipoprotein AI as a major constituent of a human hereditary amyloid. Biochem Biophys Res Commun 156:762–768
Ostertag B (1950) Familiere amyloid-erkrankung. Zeitschrift für Menschliches Vererbungs und Konstitutionslehre 30:105–115
Serpell LC, Benson M, Liepnieks JJ, Fraser PE (2007) Structural analyses of fibrinogen amyloid fibrils. Amyloid 14:199–203
Uemichi T, Liepnieks JJ, Benson MD (1994) Hereditary renal amyloidosis with a novel variant fibrinogen. J Clin Invest 93:731–736
Uemichi T, Liepnieks JJ, Alexander F, Benson MD (1996) The molecular basis of renal amyloidosis in Irish–American and Polish–Canadian kindreds. QJM 89:745–750
Uemichi T, Liepnieks JJ, Yamada T, Gertz MA, Bang N, Benson MD (1996) A frame shift mutation in the fibrinogen A alpha chain gene in a kindred with renal amyloidosis. Blood 87:4197–4203
Uemichi T, Liepnieks JJ, Gertz MA, Benson MD (1998) Fibrinogen A alpha chain Leu 554: an African–American kindred with late onset renal amyloidosis. Amyloid 5:188–192
Zeldenrust S, Gertz M, Uemichi T, Bjornsson J, Wiesner R, Schwab T, Benson M (2003) Orthotopic liver transplantation for hereditary fibrinogen amyloidosis. Transplantation 75:560–561
Acknowledgements
This project was supported by grants of the European Union (EU FP6 EURAMY). We wish to thank Dr. August (Münster), Prof. Krenn (Trier) and Prof. Waldherr (Heidelberg) for kindly providing the kidney biopsies for immunohistochemical analyses, Dr. Thomas Ackermann (Mechernich) and Dr. Kerstin Traser (Halle/Saale) for admission of the patients and Prof. Luft (Berlin) for his critical revision of the manuscript.
Conflict of interest statement
We declare that we have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eriksson, M., Schönland, S., Bergner, R. et al. Three German fibrinogen Aα-chain amyloidosis patients with the p.Glu526Val mutation. Virchows Arch 453, 25–31 (2008). https://doi.org/10.1007/s00428-008-0619-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-008-0619-4