
Abstract
It has been more than 10 years since follicular dendritic cell (FDC) sarcoma was first reported to occur in extranodal sites, yet extranodal FDC sarcoma still appears underrecognized, and its clinical and pathological characteristics remain to be defined. This study analyzed the clinical and pathological findings of three such cases that the authors encountered recently and 43 previously reported cases identified in the literature. Assessment of all 46 cases showed a slight female predominance (1.2:1) with a median age of 41.5 years. One-third of the cases were misdiagnosed at initial evaluation mainly because the possibility of FDC sarcoma was not considered. When considered, this disease had distinct pathological characteristics that allowed an accurate diagnosis. Staining for FDC markers, CD21, CD35, and clusterin was particularly helpful. The pathogenesis of the disease appeared heterogeneous, and associated factors included Epstein–Barr virus infection (in hepatic cases) and inflammatory pseudotumor-like conditions. Treatment modality varied widely although surgical resection was often included. With a median follow-up of 18 months, 43% of the cases recurred and 7% died of disease. The 5-year recurrence-free survival was 27.4%. From data available at the current time, we were not able to identify prognostically significant pathologic factors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Follicular dendritic cells (FDCs) belong to the accessory immune system. They normally reside in the primary and secondary lymphoid follicles, serving as antigen-presenting cells and playing a major role in the induction and maintenance of humoral immune response [19, 46].
Tumors showing features of FDCs were first recognized in 1986 by Monda et al. [30] who described four cases. Since then, many reports have documented FDC sarcomas, predominantly arising in lymph node [9, 22, 34]. In general, nodal FDC sarcomas tend to affect axillary or cervical lymph nodes of young to middle-aged adults without sex predilection. The behavior is believed to be more akin to that of a low-grade sarcoma than a malignant lymphoma [9, 34]. As the morphological features of FDC sarcomas (storiform arrangement of plump spindle cells with admixed lymphocytes) have become better known, the possibility of this diagnosis is now commonly being considered for node-based spindle cell lesions, and the availability of antibodies that recognize FDCs (CD21, CD35, and clusterin) has allowed the diagnosis to be established more readily.
FDC sarcoma of extranodal sites, on the other hand, appears much less well recognized, although its occurrence has been known since the early 1990s [8]. In these locations, FDC sarcoma does not usually enter the differential diagnosis clinically or pathologically, particularly when it occurs in visceral organs, and the morphological features suggest a wide variety of other diagnoses such as carcinoma, meningioma, malignant fibrous histiocytoma, and schwannoma. We have recently encountered three cases of extranodal FDC sarcoma, two involving intraabdominal sites and one involving the tonsil. All three were misdiagnosed at the time of initial evaluation, as the possibility of FDC sarcoma was not entertained in any of them. This prompted us to perform a detailed analysis of extranodal examples of this disease based on these cases as well as other well-characterized, previously reported cases. We identified 43 previously reported cases with adequate information. In this study, we detail the features of extranodal FDC sarcoma, providing an overview of the current knowledge about the clinical, pathological and histogenetic characteristics of this disease entity.
Materials and methods
Assessment of new cases
For the three new cases, a detailed clinical history was obtained from the medical records and from discussions with the responsible clinicians. Pathological materials were processed in a routine manner with formalin fixation and paraffin embedding for H&E and immunohistochemical staining. Antibodies used for immunohistochemistry included vimentin (Dako, CA, Carpinteria, CA, USA; 1:4000), CD21 (Dako, 1:10), CD35 (Dako, 1:100), clusterin (Upstate Biotech, CA, USA 1:200), AE1:AE3 (Boehringer-Mannheim, Indianapolis, IN, USA, monoclonal, 1:1,000), CAM5.2 (Becton Dickinson, Franklin Lakes, NJ, USA, monoclonal, 1:1,000), 34BE12 (Enzo, Farmingdale, NY, USA, 1:50), S100 protein (Dako, 1:500), HMB45 (Dako, 1:10), A103 (Ventana Medical Systems, prediluted), tyrosinase (Novocastra, Newcastle upon Tyne, UK, 1:200), LCA (Dako, 1:1,000), SMA (Dako, 1:1,000), Desmin (Dako, 1:200), and c-kit (Dako, 1:100). Appropriate positive and negative controls were evaluated simultaneously. Electron microscopy was performed in one case, for which ultrathin sections were cut from epoxy resin-embedded, 2% glutaraldehyde-fixed, and 1% osmium tetroxide postfixed tissues. These sections were stained with uranyl acetate and lead citrate and examined with a Zeiss 906 transmission electron microscope. In situ hybridization for Epstein–Barr virus (EBV)-encoded RNA (EBER) was performed in one case on formalin-fixed, paraffin-embedded tissue using a fluoresceinated EBV-specific RNA probe (Dako). Positive signal was detected with a biotin-free, antifluorescein horseradish peroxidase-conjugated antibody.
Literature review
Review of the literature was performed with a MEDLINE search using the terms “follicular dendritic cell tumor” or “follicular dendritic cell sarcoma” combined with “extranodal” or with specific sites (e.g., liver or hepatic, stomach or gastric, etc). References from articles retrieved by this method were also reviewed to identify other relevant publications. An effort was made to identify cases that were reported more than once in different settings, and only one entry with the most updated information was included for such cases. Nodal and splenic FDC tumors were not included.
Statistical analysis
Data obtained from the literature and the three new cases were analyzed. Recurrence-free survival rates were calculated by Kaplan–Meier estimation. Associations between pathological factors and adverse outcome were analyzed using the Cox regression method.
Results
Clinical and pathological findings of the three new cases
Pertinent clinical data on all three cases are listed in Table 1. In case 1, the disease occurred in the rectum of a 30-year-old male patient. The patient presented with a 6-month history of blood per rectum, and an endoscopy showed a mass protruding into the rectal lumen. The initial biopsy of the mass was thought to be a “mesenchymal tumor with neural differentiation”. The patient underwent a low anterior resection and opted not to receive any adjuvant therapy postoperatively. Sixteen months after the diagnosis, a follow-up CT scan demonstrated perihepatic intraperitoneal masses consistent with recurrent disease. No further workup or treatment was carried out at the time of last follow-up, which was 19 months after the initial diagnosis.
Case 2 was a 29-year-old female patient who presented with a mass in the left upper quadrant of the abdomen. A workup including ultrasound, CT scan, and MRI revealed a mass in close proximity to the stomach and liver. The mass was resected without a preoperative biopsy. At operation, the 12.0×9.0×4.0-cm mass appeared to have arisen from the lesser omentum, adjacent but not adherent to the neighboring organs. The pathological diagnosis rendered for this tumor at the referring institution was “a high-grade neoplasm, possibly a malignant fibrous histiocytoma, giant cell type”. Because of the presence of giant cells and close proximity to the pancreas, a concern of “a sarcomatoid giant cell carcinoma of the pancreas” was also raised. At last follow-up, 17 months after initial diagnosis, the patient remained alive with no evidence of disease; no additional therapy was given.
Case 3 involved the tonsil of a 69-year-old female patient. Pathological diagnosis for the initial excisional biopsy was “squamous cell carcinoma of the tonsil”, and the patient underwent adjuvant radiation therapy followed by a radical neck dissection. The patient had then remained disease-free for 8 years, at which point she developed a persistent cough and was found to have hilar and lung lesions. A mediastinoscopic biopsy of the hilar lesion showed a metastatic FDC sarcoma involving a lymph node. Retrospective review of the patient’s prior tonsillar lesion revealed an FDC sarcoma as well. No further therapy was given. At last follow-up, 9 years after the initial tonsillar lesion, the patient was alive with radiographic evidence of stable disease.
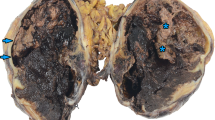
Pathological features of all three cases are summarized in Table 2. Grossly, the tumors were well circumscribed with a fleshy cut surface, similar to sarcoma or lymphoma (Fig. 1). Microscopically, cases 1 and 2 showed a more spindled cell morphology with a whorled, fascicular, or storiform growth pattern (Fig. 2), while in case 3, the cells were more epithelioid and the growth pattern was more diffuse (Fig. 3). In all three cases, the cells had eosinophilic fibrillary cytoplasm with indistinct cell borders (Fig. 4), and there were small lymphocytes scattered throughout the tumor and clustered around vessels (Fig. 5). In case 3, the epithelioid tumor cells had irregular nuclei and prominent round nucleoli. Some nuclei contained round intranuclear inclusions (Fig. 3). In all three cases, mitoses were not prominent, being absent in two cases and 3/10 high power fields (HPF) in one (case 1). Necrosis was noted in two cases. Regional lymph node involvement was present in case 1. Immunohistochemically, all three cases showed diffuse positivity for CD21, two of three showed positivity for CD35, and one of one for clusterin (Fig. 6). All cases were negative for cytokeratins, and two of three were negative for S100 protein. Cases 1 and 2 were tested for c-kit, and muscle markers (desmin and SMA) with negative results. Melanoma markers, HMB45 and tyrosinase, were negative in all three. Ultrastructural studies were performed in case 1, as was in situ hybridization for EBER. Ultrastructurally, the tumor cells showed prominent cell processes that were focally joined by well-formed cell junctions and desmosomes (Fig. 7). EBER showed no evidence of EBV.

Gross picture of an FDC sarcoma of the rectum (case 1). There are two main mass lesions, both affecting the bowel wall. Note the fleshy homogeneous cut surface with focal hemorrhage and necrosis

Microscopic appearance of an FDC sarcoma of the rectum (case 2). The tumor has a whorled and storiform growth pattern. The tumor cells are spindled

An FDC sarcoma of the tonsil (case 3) showing a diffuse growth pattern. The tumor cells are plump and epithelioid with pink, vaguely fibrillary cytoplasm. The nuclei are irregular and cleaved in some cells. Prominent nucleoli are present. Some nuclei contain round intranuclear inclusions. Multinucleated tumor giant cells are also apparent

The tumor cells of an FDC sarcoma showing eosinophilic fibrillary cytoplasm with indistinct cell borders (case 1)

FDC sarcoma showing small lymphocytes scattered about and clustered around blood vessels (case 1)

FDC sarcoma showing diffuse positivity to clusterin by immunohistochemistry (case 1)

Ultrastructurally, the tumor cells show prominent cell processes that are focally joined by well-formed cell junctions and desmosomes (case 1)
Literature review
A total of 43 cases of extranodal FDC sarcoma were retrieved from the literature. Clinical information on these 43 cases, along with the three new cases, is detailed in Table 1.
Patient demographic characteristics
There were 25 women and 21 men (female to male ratio, 1.2:1). The age range was 9–82 years, with a median of 41.5 years and a mean of 43.5 years. The geographic distribution of the cases covered both Eastern and Western countries, although more reports appeared to have originated from Asian countries.
Tumor anatomic distribution
The tumors affected various anatomic sites. Six involved the liver. Another six involved the tubular gastrointestinal tract (three in large bowel, one in small bowel, and two in stomach). Other organ sites included the ampulla of Vater, pancreas, lung, breast, and thyroid (one case each). Three cases were intraabdominal but extravisceral (one in lesser omentum, one in mesocolon, and one in retroperitoneum). Eight cases involved soft tissues of the neck (five cases), the abdominal wall (two cases), and the thigh (one case). Seventeen cases involved the oral cavity and upper aerodigestive regions (ten in tonsil, seven in palate and pharynx) and one the parotid gland. Of note, the three cases that were intraabdominal and extravisceral, and the eight cases of soft tissues of various sites were included as extranodal FDC tumors based on lack of convincing evidence of nodal origin.
Clinical management and patients’ outcome
Eight of the 46 cases (17%) had lymph node involvement at the time of presentation, and one of the eight also had a liver metastasis. Except in two cases (cases 6 and 32), all patients underwent surgical resection of the tumor. Eighteen of the 44 patients (41%) who had surgery also had some form of adjuvant treatment: 4 had neoadjuvant chemotherapy, 3 had adjuvant chemotherapy, 2 had neoadjuvant radiotherapy (RT), 8 had adjuvant RT, and 1 had adjuvant chemo and RT. The chemotherapeutic agents varied among reports but usually included the cytoxan, hydroxydaunomycin (adriamycin), vincristine (oncovin), and prednisone (CHOP) regimen.
Follow-up information was available in 42 cases. The follow-up time ranged from 5 to 324 months (mean, 32 months; median, 18 months). Overall, 18 of the 42 patients (43%) had local and/or distant recurrence after initial treatment. Local recurrence occurred in 11 patients (26%) at a median of 15 months after resection (range, 6–60 months). Distant metastases occurred in nine patients (21%) at a median of 24 months after resection (range, 6–324 months). Metastatic sites included lung (five cases), liver (one case), intraabdominal soft tissues (one case), and lymph nodes (two cases). Two cases (cases 20 and 27) had both local and distant recurrence. At last follow-up, three patients (7%) died of disease, 15 (36%) were alive with disease, and 24 (57%) were alive with no evidence of disease. Two-year and 5-year recurrence-free survival for the entire group were 62.3% (95% CI, 0.469, 0.827) and 27.4% (95% CI, 0.102, 0.739), respectively (Fig. 8).

Recurrence-free survival curve for patients with extranodal FDC sarcoma
Discordant diagnoses entertained at initial evaluation
Overall, 15 cases (33%) were misdiagnosed at the time of initial evaluation. In all three new cases, and in some reported cases, the diagnosis of FDC sarcoma was not entertained at initial evaluation. In some cases, such erroneous interpretations were the reasons that neoadjuvant or adjuvant therapy was rendered. Disease entities considered included: reactive response (one case), carcinoma (five cases), inflammatory pseudotumor (IPT) (two cases), malignant fibrous histiocytoma (two cases), meningioma (one case), mesenchymal tumor with neural differentiation (or neural origin) (two cases), schwannoma (one case), and stromal tumor (one case).
Pathological findings
The average size of tumors of all sites was 7.4 cm, ranging from 1 to 20 cm (median, 6 cm). Intraabdominal tumors (n=18) had an average tumor size of 10.3 cm (range, 4–20 cm; median, 11 cm), whereas the average tumor size of the tumors outside the abdominal cavity (n=19) was only 4.7 cm (range, 1–14 cm; median, 3 cm) (p<0.001).
The gross and microscopic findings of the reported cases were similar to those observed in our three cases as described above. The most helpful diagnostic features were the syncytial arrangement of tumor cells with fibrillary cytoplasm and indistinct cytoplasmic borders and the presence of small lymphocytes scattered throughout the tumor. The cells varied from plump and spindled to epithelioid. It is of interest to note that FDC sarcomas arising in the liver tended to have a prominent plasma cell component in the inflammatory infiltrate, whereas plasma cells were nonexistent or inconspicuous in extrahepatic cases. Binucleated and multinucleated neoplastic cells and intranuclear inclusions were features that were seen in some cases. Although the tumors were often hypercellular, most reports suggested that the tumors had a low-grade histology with only mild to moderate atypia. However, high-grade examples with marked cytological atypia were reported. A mitotic count was available in 31 cases, ranging from 0 to 50/10 HPF with a median of 3/10 HPF. Nineteen of 31 (61%) had a mitotic count of <5/10 HPF. Information on necrosis was available in 33 cases, of which ten (30%) exhibited coagulative necrosis.
Immunohistochemical staining results are summarized in Table 3. CD21 and CD35 were commonly used, and both had a high positive rate (98% for CD21 and 93% for CD35). Clusterin and fascin were relatively recently employed markers [22, 23] and were tested in only a few cases, having uniform positivity. CD68, S100 protein, and EMA showed variable staining results among different reports. Desmoplakin, a desmosome-associated protein [17], was detected in 73% of the cases, while cytokeratin was positive in only 1 of 18 cases. In the one cytokeratin-positive case, the positivity was only seen in <10% of the tumor cells.
Ultrastructural findings were reported in ten cases, and all showed typical findings as described in our case 1.
Correlation between clinical and pathological findings and patients’ outcome
Associations between the various clinical and pathological factors and recurrence-free survival were analyzed, and the results are summarized in Table 4. Factors assessed included age, gender, intraabdominal involvement, size, lymph node involvement or distant metastasis at time of presentation, mitotic rate, necrosis, and the use of neoadjuvant or adjuvant therapy. No statistical significance was identified for any of these factors.
Possible etiology and associated conditions
Of the six tumors that occurred in the liver, five were positive for the EBER as tested by in situ hybridization. Furthermore, Southern blot analysis in two cases showed clonal and episomal EBV in the tumor cells [41]. On the other hand, all 24 extra-hepatic cases that were tested by EBER failed to demonstrate any evidence of EBV.
In addition, two cases (cases 16 and 37) [7, 9] had an association with Castleman’s disease of hyaline-vascular type [9]. In case 16, the main tumor mass showed small foci of residual hyaline-vascular Castleman’s disease [9]. In case 37, hyaline-vascular Castleman’s disease occurred at the same site 11 years before development of a frank FDC sarcoma [7]. Another two cases (cases 1 and 5), both occurring in the liver, exhibited a morphological resemblance to the so-called IPT and were diagnosed as such initially [39, 41].
Discussions
Given the fact that FDCs are present in all lymphoid follicles, including extranodal lymphoid follicles, it is not surprising that tumors of these cells occur in extranodal sites. Yet, extranodal FDC sarcoma appears to be significantly underrecognized, particularly when it affects visceral organs. This entity did not enter the differential diagnosis at the time of initial evaluation in any of the three cases we encountered.
When suspected based on histology, the diagnosis of FDC sarcoma can be relatively straightforward. As illustrated by this review study, extranodal FDC sarcomas, like their nodal counterparts, typically have distinct histological, immunophenotypical, and ultrastructural findings to allow a definitive diagnosis. In terms of immunohistochemical markers, in addition to CD21 and CD35, clusterin has been shown to be a highly sensitive and specific FDC marker [23]. Fascin, another relatively recently discovered marker, however, has been shown to be very nonspecific among spindle cell tumors and thus does not imply a dendritic lineage [23]. Desmoplakin is a desmosome-associated protein found on epithelial cells and nonepithelial cells with desmosomes [17]. Positive staining was detected in 73% of the cases. However, because the antibody does not yield consistent results on paraffin-embedded sections, often producing only patchy or focal staining, it is of limited value for the diagnosis of FDC sarcoma. Additionally, FDCs may show variable staining for EMA, S100 protein, and CD68. Keratin stains, however, appear consistently negative in FDC sarcoma; in only 1 of the 18 cases tested was there focal keratin staining in <10% of the tumor. These immunohistochemical results for extranodal FDCs are essentially identical to the more extensive results reported for nodal FDCs.
It is difficult to determine the frequency of extranodal FDC sarcoma given that almost all reports are case-based and the denominator necessary for the determination of the frequency is essentially unknown. Overall, the disease is uncommon. Lack of a high index of suspicion is the main reason for this uncommon disease to be misdiagnosed in most instances, as illustrated in all three new cases we encountered and those reported in the literature. When evidence of FDC differentiation is not sought, the fact that FDC sarcomas can show variable staining for EMA, CD68, and S100 protein, coupled with an epithelioid cell-predominant or a spindle cell-predominant morphology, may lead to such diagnoses as carcinoma, meningioma, sarcoma of neural origin, malignant fibrous histiocytoma, or simply a stromal tumor not further specified. As documented in case 3 of our series and several cases reported in the literature, such erroneous diagnoses have then led to divergent treatment modalities for this disease entity.
Correct identification of this disease entity is clinically important. As our analyses have shown, this disease carries an overall recurrence rate of 43% (local recurrence in 26% and distant recurrence in 21%). The 5-year recurrence-free survival rate was only 27.4%. A small but definite proportion of patients (7%) died of the disease. These figures may still underestimate the biology of this tumor given the relatively short follow-up time available for these cases.
How to predict which patients will experience a worse outcome is a question still, however, to be answered. Previous reports suggested that intraabdominal involvement, a high mitotic count (≥5/10 HPF), coagulative necrosis, and significant cellular atypia are potentially helpful morphological predictors of unfavorable outcome [9]. In this study, we were not able to perform a statistical analysis on cellular atypia because data were not uniformly recorded in the literature reports. However, none of the other factors mentioned above showed significant statistical power in predicting tumor recurrence. With increasing recognition of FDC sarcomas, cases with more marked nuclear pleomorphism are being diagnosed more frequently, and it remains to be established whether a more poorly differentiated FDC sarcoma may also be more clinically aggressive. Similarly, more experience is needed to determine any other prognostic factors.
With regard to the etiology and predisposing factors for FDC sarcoma, there have been a number of interesting observations that deserve attention and further investigation. First, as outlined in this review study, five of six hepatic FDCs exhibited positive EBER by in situ hybridization, whereas none of the 24 extrahepatic cases tested was positive for the virus. EBV has also been demonstrated in some but not all splenic and nodal FDC sarcomas [6, 26]. In the hepatic cases, Southern blotting analysis showed that the EBV infection occurred before the clonal proliferation of neoplastic FDCs, suggesting its causative role in the development of FDC sarcoma [39, 41]. These findings therefore suggest a pathogenetic difference between hepatic (and some splenic and nodal) FDC sarcomas and other types of FDC sarcomas.
Second, as illustrated by cases 17 and 37, some extranodal FDC sarcomas appear to be associated with the hyaline-vascular type Castleman’s disease, similar to some of their nodal counterparts [7, 27, 28]. This finding is not surprising because Castleman’s disease affects extranodal sites as well. Several reports have suggested that the FDC proliferation and dysplastic changes occurring in Castleman’s disease can form the background from which an FDC sarcoma develops. As such, the relationship of Castleman’s disease and FDC sarcomas has been suggested to parallel the hyperplasia–dysplasia–neoplasia sequence that is seen in other tumor types [9, 29, 35, 36].
Third, there have been emerging data suggesting an association between FDC sarcoma and the so-called IPT [6, 12, 32, 39, 41]. In this study, IPT refers to the morphologically reactive fibroinflammatory mass lesions of uncertain nature and etiology. Such an association appears to be particularly apparent in hepatic cases. Two of the reported hepatic cases (cases 4 and 8) were initially diagnosed as IPT. More importantly, Cheuk et al. [12] in 2001 reported a series of seven hepatic, three splenic, and one peripancreatic FDC proliferations that bore a “striking histological resemblance to IPT because of the admixture of lymphocytes, plasma cells, and spindle cells”. Although only one splenic case showed a component that was indistinguishable morphologically from a conventional FDC tumor, the authors believed that such FDC proliferations were neoplastic. Cossu et al. [15] in 2005 reported a classic FDC tumor developing in a 31-year-old woman, at a site where a nodal IPT-like proliferation was removed 6 years earlier. It is of great interest that in the initial IPT-like lesion, groups of irregularly and loosely arranged CD21-positive cells were noted. All these observations suggest that either FDC sarcoma can present at a very early stage as an IPT-like condition or a subset of IPT has the potential to evolve into an FDC sarcoma. Of interest also is the fact that all hepatic cases [39, 41, 42] (including that reported by Cheuk et al. [12]) exhibited EBV genome in the tumor cells whereas the nodal FDC sarcoma that developed at the site of a previously excised IPT did not [15].
Viewing the above observations together, it appears that FDC sarcomas, nodal or extranodal, are pathogenetically heterogeneous. Most hepatic FDC sarcomas and some splenic and nodal tumors might be related to EBV infection and might present as a fibroinflammatory mass like the so-called IPT. The cases reported by Cheuk et al. [12] might represent an earlier stage of FDC sarcoma when compared to those reported by others [11, 39, 41]. All these cases seem to share a heavy inflammatory infiltrate that may contain plasma cells. Most nonhepatic FDC sarcomas, on the other hand, may have an inciting factor not related to EBV [15]. Some such cases might also undergo an IPT-like proliferation at an early stage or might have evolved from an IPT-like condition.
It is apparent that much more investigation is required, including perhaps clonality studies, before we can firmly accept the pathogenetic relationship between some IPT-like conditions and FDC sarcoma. The association between IPT and FDC sarcoma, however, is not unreasonable from a conceptual point of view and has already been suggested by other investigators [32]. After all, FDCs are believed to be of mesenchymal origin. Phenotypic marker studies and in vitro experiments with fibroblast-like cell lines have demonstrated development of FDCs from fibroblast-like cells [4, 44]. It is possible that under certain oncogenic stimuli, such as EBV infection, the mesenchymal cells in IPT-like conditions undergo transformation into FDCs, and as has been observed in Castleman’s disease, such cells then undergo “dysplasia” and eventually become neoplastic.
In summary, this study illustrates the need for wider recognition of extranodal FDC sarcoma. In analyzing all reported cases, we have outlined the clinical and pathological characteristics of this disease entity and brought forth evidence for associations between FDC sarcoma and IPT-like conditions which should prompt further exploration of the histogenesis and classification of both IPT and FDC sarcoma.
References
Araujo VC, Martins MT, Salmen FS, Araujo NS (1999) Extranodal follicular dendritic cell sarcoma of the palate. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 87:209–214
Beham-Schmid C, Beham A, Jakse R, Aubock L, Hofler G (1998) Extranodal follicular dendritic cell tumour of the nasopharynx. Virchows Arch 432:293–298
Biddle DA, Ro JY, Yoon GS, Yong YW, Ayala AG, Ordonez NG, Ro J (2002) Extranodal follicular dendritic cell sarcoma of the head and neck region: three new cases, with a review of the literature. Mod Pathol 15:50–58
Bofill M, Akbar AN, Amlot PL (2000) Follicular dendritic cells share a membrane-bound protein with fibroblasts. J Pathol 191:217–226
Bradshaw EJ, Wood KM, Hodgkinson P, Lucraft H, Windebank KP (2005) Follicular dendritic cell tumour in a 9-year-old child. Pediatr Blood Cancer 45:725–727
Brittig F, Ajtay E, Jakso P, Kelenyi G (2004) Follicular dendritic reticulum cell tumor mimicking inflammatory pseudotumor of the spleen. Pathol Oncol Res 10:57–60
Chan AC, Chan KW, Chan JK, Au WY, Ho WK, Ng WM (2001) Development of follicular dendritic cell sarcoma in hyaline-vascular Castleman’s disease of the nasopharynx: tracing its evolution by sequential biopsies. Histopathology 38:510–518
Chan JK, Tsang WY, Ng CS, Tang SK, Yu HC, Lee AW (1994) Follicular dendritic cell tumors of the oral cavity. Am J Surg Pathol 18:148–157
Chan JK, Fletcher CD, Nayler SJ, Cooper K (1997) Follicular dendritic cell sarcoma. Clinicopathologic analysis of 17 cases suggesting a malignant potential higher than currently recognized. Cancer 79:294–313
Chang KC, Jin YT, Chen FF, Su IJ (2001) Follicular dendritic cell sarcoma of the colon mimicking stromal tumour. Histopathology 38:25–29
Chen TC, Kuo TT, Ng KF (2001) Follicular dendritic cell tumor of the liver: a clinicopathologic and Epstein–Barr virus study of two cases. Mod Pathol 14:354–360
Cheuk W, Chan JK, Shek TW, Chang JH, Tsou MH, Yuen NW, Ng WF, Chan AC, Prat J (2001) Inflammatory pseudotumor-like follicular dendritic cell tumor: a distinctive low-grade malignant intra-abdominal neoplasm with consistent Epstein–Barr virus association. Am J Surg Pathol 25:721–731
Chiaramonte MF, Lee D, Abruzzo LV, Heyman M, Bass BL (2001) Retroperitoneal follicular dendritic cell sarcoma presenting as secondary amyloidosis. Surgery 130:109–111
Choi PC, To KF, Lai FM, Lee TW, Yim AP, Chan JK (2000) Follicular dendritic cell sarcoma of the neck: report of two cases complicated by pulmonary metastases. Cancer 89:664–672
Cossu A, Lissia A, Dedola MF, Deiana A, Faedda R, Palmieri G, Tanda F (2005) Classic follicular dendritic reticulum cell tumor of the lymph node developing in a patient with a previous inflammatory pseudotumor-like proliferation. Hum Pathol 36:207–211
Desai S, Deshpande RB, Jambhekar N (1999) Follicular dendritic cell tumor of the parapharyngeal region. Head Neck 21:164–167
Ennas MG, Chilosi M, Scarpa A, Lantini MS, Cadeddu G, Fiore-Donati L (1989) Isolation of multicellular complexes of follicular dendritic cells and lymphocytes: immunophenotypical characterization, electron microscopy and culture studies. Cell Tissue Res 257:9–15
Fisher C, Magnusson B, Hardarson S, Smith ME (1999) Myxoid variant of follicular dendritic cell sarcoma arising in the breast. Ann Diagn Pathol 3:92–98
Fonseca R, Yamakawa M, Nakamura S, van Heerde P, Miettinen M, Shek TW, Myhre Jensen O, Rousselet MC, Tefferi A (1998) Follicular dendritic cell sarcoma and interdigitating reticulum cell sarcoma: a review. Am J Hematol 59:161–167
Galati LT, Barnes EL, Myers EN (1999) Dendritic cell sarcoma of the thyroid. Head Neck 21:273–275
Geerts A, Lagae E, Dhaene K, Peeters M, Waeytens A, Demetter P, Cuvelier C, Defreyne L, De Vos M, Pattyn P (2004) Metastatic follicular dendritic cell sarcoma of the stomach: a case report and review of the literature. Acta Gastroenterol Belg 67:223–227
Grogg KL, Lae ME, Kurtin PJ, Macon WR (2004) Clusterin expression distinguishes follicular dendritic cell tumors from other dendritic cell neoplasms: report of a novel follicular dendritic cell marker and clinicopathologic data on 12 additional follicular dendritic cell tumors and 6 additional interdigitating dendritic cell tumors. Am J Surg Pathol 28:988–998
Grogg KL, Macon WR, Kurtin PJ, Nascimento AG (2005) A survey of clusterin and fascin expression in sarcomas and spindle cell neoplasms: strong clusterin immunostaining is highly specific for follicular dendritic cell tumor. Mod Pathol 18:260–266
Han JH, Kim SH, Noh SH, Lee YC, Kim HG, Yang WI (2000) Follicular dendritic cell sarcoma presenting as a submucosal tumor of the stomach. Arch Pathol Lab Med 124:1693–1696
Hollowood K, Stamp G, Zouvani I, Fletcher CD (1995) Extranodal follicular dendritic cell sarcoma of the gastrointestinal tract. Morphologic, immunohistochemical and ultrastructural analysis of two cases. Am J Clin Pathol 103:90–97
Horiguchi H, Matsui-Horiguchi M, Sakata H, Ichinose M, Yamamoto T, Fujiwara M, Ohse H (2004) Inflammatory pseudotumor-like follicular dendritic cell tumor of the spleen. Pathol Int 54:124–131
Katano H, Kaneko K, Shimizu S, Saito T, Irie T, Mori S (1997) Follicular dendritic cell sarcoma complicated by hyaline-vascular type Castleman’s disease in a schizophrenic patient. Pathol Int 47:703–706
Lee IJ, Kim SC, Kim HS, Bang D, Yang WI, Jung WH, Chi HS (1999) Paraneoplastic pemphigus associated with follicular dendritic cell sarcoma arising from Castleman’s tumor. J Am Acad Dermatol 40:294–297
Lin O, Frizzera G (1997) Angiomyoid and follicular dendritic cell proliferative lesions in Castleman’s disease of hyaline-vascular type: a study of 10 cases. Am J Surg Pathol 21:1295–1306
Monda L, Warnke R, Rosai J (1986) A primary lymph node malignancy with features suggestive of dendritic reticulum cell differentiation. A report of 4 cases. Am J Pathol 122:562–572
Nayler SJ, Verhaart MJ, Cooper K (1996) Follicular dendritic cell tumour of the tonsil. Histopathology 28:89–92
Nonaka D, Birbe R, Rosai J (2005) So-called inflammatory myofibroblastic tumour: a proliferative lesion of fibroblastic reticulum cells? Histopathology 46:604–613
O’Malley D (2004) Diagnosis: follicular dendritic cell tumor mimicking GI stromal tumor. http://socforheme.org/case-nov-04.htm
Perez-Ordonez B, Erlandson RA, Rosai J (1996) Follicular dendritic cell tumor: report of 13 additional cases of a distinctive entity. Am J Surg Pathol 20:944–955
Perez-Ordonez B, Rosai J (1998) Follicular dendritic cell tumor: review of the entity. Semin Diagn Pathol 15:144–154
Ruco LP, Gearing AJ, Pigott R, Pomponi D, Burgio VL, Cafolla A, Baiocchini A, Baroni CD (1991) Expression of ICAM-1, VCAM-1 and ELAM-1 in angiofollicular lymph node hyperplasia (Castleman’s disease): evidence for dysplasia of follicular dendritic reticulum cells. Histopathology 19:523–528
Satoh K, Hibi G, Yamamoto Y, Urano M, Kuroda M, Nakamura S (2003) Follicular dendritic cell tumor in the oro-pharyngeal region: report of a case and a review of the literature. Oral Oncol 39:415–419
Schwarz RE, Chu P, Arber DA (1999) Extranodal follicular dendendritic cell tumor of the abdominal wall. J Clin Oncol 17:2290–2292
Selves J, Meggetto F, Brousset P, Voigt JJ, Pradere B, Grasset D, Icart J, Mariame B, Knecht H, Delsol G (1996) Inflammatory pseudotumor of the liver. Evidence for follicular dendritic reticulum cell proliferation associated with clonal Epstein–Barr virus. Am J Surg Pathol 20:747–753
Shah RN, Ozden O, Yeldandi A, Peterson L, Rao S, Laskin WB (2001) Follicular dendritic cell tumor presenting in the lung: a case report. Hum Pathol 32:745–749
Shek TW, Ho FC, Ng IO, Chan AC, Ma L, Srivastava G (1996) Follicular dendritic cell tumor of the liver. Evidence for an Epstein–Barr virus-related clonal proliferation of follicular dendritic cells. Am J Surg Pathol 20:313–324
Shek TW, Liu CL, Peh WC, Fan ST, Ng IO (1998) Intra-abdominal follicular dendritic cell tumour: a rare tumour in need of recognition. Histopathology 33:465–470
Torres U, Hawkins WG, Antonescu CR, DeMatteo RP (2005) Hepatic follicular dendritic cell sarcoma without Epstein–Barr virus expression. Arch Pathol Lab Med 129:1480–1483
Van Nierop K, de Groot C (2002) Human follicular dendritic cells: function, origin and development. Semin Immunol 14:251–257
Vargas H, Mouzakes J, Purdy SS, Cohn AS, Parnes SM (2002) Follicular dendritic cell tumor: an aggressive head and neck tumor. Am J Otolaryngol 23:93–98
Wu J, Qin D, Burton GF, Szakal AK, Tew JG (1996) Follicular dendritic cell-derived antigen and accessory activity in initiation of memory IgG responses in vitro. J Immunol 157:3404–3411
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shia, J., Chen, W., Tang, L.H. et al. Extranodal follicular dendritic cell sarcoma: clinical, pathologic, and histogenetic characteristics of an underrecognized disease entity. Virchows Arch 449, 148–158 (2006). https://doi.org/10.1007/s00428-006-0231-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-006-0231-4