
Abstract
While diffuse mesangial sclerosis is traditionally described as being the glomerulopathy of Denys–Drash syndrome (DDS), the podocyte proliferative lesions may be overlooked in these DDS cases. In the present study, an evolving process is extrapolated from a selected case of DDS that demonstrated glomerulopathy with conspicuous podocyte proliferation. The observation that podocytes express proliferation markers (Ki67, proliferating-cell nuclear antigen and topoisomerase IIα) in non-proliferative, mature-looking glomeruli suggests an initial pathogenic act to activate or to keep podocytes from quiescence. The subsequent proliferation of podocytes is in keeping with downregulation of WT1 and cyclin kinase inhibitors of p16 and p21. The emergence of cytokeratin-positive cells in glomeruli that show typical mesangial sclerosis implies elimination of podocytes and replacement with tubular and/or parietal epithelial cells. The final scene of evolving glomerulopathy displays apoptosis and expression of Fas-L and Bax in sclerotic mesangial lesions, which eventually end up with global sclerosis. This novel concept of DDS glomerulopathy implies complex molecular mechanisms involved in glomerular injury.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The renal involvement in the Denys–Drash syndrome (DDS) is manifested by Wilms’ tumor and glomerulopathy. The latter eventually leads to end-stage renal failure and is the primary cause of death in this syndrome [19]. The investigation of genetic abnormality in nearly all patients of DDS reveals constitutionally heterozygous point mutations for WT1, a gene that encodes a transcription factor that functions in kidney and gonad proliferation and differentiation [2, 16, 33]. Incomplete DDS and isolated diffuse mesangial sclerosis (IDMS), which share similar glomerulopathy with the complete form of DDS, also demonstrate various wt1 mutations [33]. It has been suggested that glomerulopathy results from debilitating transcriptional function of WT1 in a dominant negative fashion [2, 16, 17]. A mouse model of DDS, which was heterozygous for a target wt1 allele, demonstrated truncated zinc finger 3 at amino acid 396 of WT1 protein and severe proliferative glomerulopathy with crescent formation [24]. In a human study, mild increase of cell proliferation was demonstrated by proliferating-cell nuclear antigen (PCNA)-positive podocytes in two-thirds of DDS patients; one case even showed abundant positive cells in up to 50% glomeruli [33]. Recent studies have revealed that some genes of the WT1 network may play pathogenic roles directly or indirectly linked to the growth control of glomerular cells. For example, PAX2, a transcriptional factor that is downregulated by WT1 during normal maturation of podocytes, is persistently expressed in podocytes of DDS and IDMS kidneys [4, 25, 33]. The PAX2 is speculated to be capable of stimulating cell proliferation by the observations that PAX2 induced podocyte hypertrophy and increased PCNA nuclear labeling [33]. Another target gene of WT1, novH, whose expression in podocytes is also downregulated by WT1 during normal nephrogenesis [18, 22], is capable of stimulating cell proliferation in synergy with growth factors such as transforming growth factor-β and basic fibroblast growth factors [6]. These data imply that the basic pathology of glomerulopathy in DDS and related disorders may be associated with dysregulation of cell proliferation in glomeruli.
The major purpose of our study was to analyze the growth response of glomerular cells in DDS kidney. We assessed cellular proliferation activity, cell-cycle regulatory factors, WT1 protein expression, apoptosis and apoptotic modulation factors to explore the pathogenic mechanism of DDS glomerulopathy.
Materials and methods
Tissue
We selected a case of DDS from our autopsy archives for this study. The clinical and histopathological features are summarized in Table 1. The major presenting symptoms and signs of this patient were a renal mass at the left kidney, nephrotic syndrome and genital abnormalities. We also retrieved control tissues of patients of similar age with neither DDS nor Wilms’ tumor or of patients with Wilms’ tumor but without DDS from surgical or autopsy archives.
Morphological studies and immunohistochemical analyses
The kidney tissues subjected to histochemical and immunohistochemical examinations were paraffin embedded and cut into 3-μm-thick sections. Sections were stained for hematoxylin and eosin, periodic acid–Schiff and periodic acid silver methanamine for routine morphological examination. The paraffin sections for immunohistochemical analyses were deparaffinized in xylene substitute (Paraclean, Earthsafe Tech, USA), rehydrated and immersed in 3% H202/methanol for 10 min. The primary antibodies used in this study are listed in Table 2. Before incubation with primary antibodies, sections were microwaved either in 0.01 M sodium citrate buffer (pH 6.0) for 10 min or with pressure cooker in 1.0 mM ethylenediaminetetraacetate buffer for 5 min and blocked with blocking serum in phosphate-buffered saline (PBS) for 20 min at room temperature. The sections were then subjected to incubation in primary antibody and peroxidase–antiperoxidase (PAP) reagents for 30 min, respectively. The sections were thoroughly washed between incubations. After the final wash in 1% Triton X-100/PBS, sections were stained using diaminobenzidine (DAB)–H2O2 as substrate. Negative controls included replacing primary antibodies with normal serum or omission of PAP reagents or both. Additional absorption control using specific blocking peptide (sc192 P, Santa Cruz) was applied to WT1 staining procedure. Appropriate tissue sections containing the specific antigen were prepared from the surgical specimen to serve as positive controls.
Assessment of apoptosis
Enzymatic in situ labeling of apoptosis induced DNA strand breaks [transferase–mediated dUTP nick end-labeling (TUNEL) assay] was assessed using the kit (#1684817 POD, Boehringer, Mannheim) as previously described with slight modification [32]. Deparaffinized sections were stained according to the instruction given by the manufacturer. Chromogenic reaction was carried out using DAB as substrate, and all sections were counterstained with hematoxylin prior to analysis under light microscope. The negative controls used label solution without terminal transferase, while the positive controls received pre-treatment of DNase I (10 μg/ml, room temperature, 10 min) prior to TUNEL reaction.
DNA sequencing
Genomic DNA was retrieved from archived paraffin blocks. The WT-1 coding sequence (exon 1 to exon 10) and flanking intron–exon boundaries were amplified using polymerase chain reaction (PCR) with a set of 12 pairs of primers (exon 1: ex1.1f 5′CGG CCC CTC TTA TTT GAG CT3′ and ex1.1r 5′AAC GAC CCG TAA GCC GAA G3′; ex1.2f 5′TCC ACG TGT GTC CCG GA3′ and ex1.2r 5′GAA AAG TGG ACA GTG AAG GCG3′; ex1.3f 5′CGC CTC ACT CCT TCA GGC TCC TA3′ and ex1.3r 5′TGC GAA CAG TCA GGC TCC TA3′; exon 2: ex2f 5′CGA GAG CAC CGC TGA CAC T3′ and ex2r 5′CTT TGC CTA ATT TGC TGT GGG3′; exon 3: ex3f 5′CAC TCG CTC AGC TGT CTT CG3′ and ex3r 5′AAC ATA AGA ACA ACT CAG AGA CCA GG3′; exon 4: ex4f 5′TGT ATG AAG TGT ATA ACT GTG CAG CAG AGA TC3′ and ex4r 5′CCA GAT AGA AAC AAC CCA CAT TTA TG3′; exon 5: ex5f 5′AAG CTC TTT GCA GCC TCC G3′ and ex5r 5′GGG CAT GTT GAT GAG AAA TGC3′; exon 6: ex6f 5′TCT AAA GTG GCC CCA TGG AG3′ and ex6r 5′CCG CCT TAT CAG ACC CAG G3′; exon 7: ex7f 5′ACA TGT GGC TTA AAG CCT CCC3′ and ex7r 5′TGA GAG CCT GGA AAA GGA GC3′; exon 8: ex8f 5′CAG CGA AGT GCC TTA GGC ATT3′ and ex8r 5′ACG CTA CAA ATT GGA TTC CGC3′; exon 9: ex9f 5′CTC TGT GGT TAA GGA ATG GTT G3′ and ex9r 5′AAC TTT TAC ACT AGT CTT TTC CAA TCC C3′; exon 10: ex10f 5′CCC AAG AGG TGC TGC TGT TT3′ and ex10r 5′AGG GCA GAG ACC AAC TCT TCC3′). All amplified regions were sequenced on both strands by means of automatic fluorescent sequencing, using the sequencing kit (Bigdye Terminator) on an ABI sequencer (3100 Genetic Analyzer). Three randomly selected parallel tissue samples from non-DDS patients were used as control.
Results
Analyses of glomerular cell proliferation
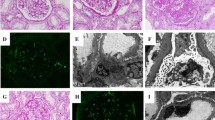
The clinical and pathological features of the DDS patient are summarized in Table 1. In the DDS kidneys, focal glomeruli show typical glomerular lesions composed of sclerotic mesangial masses surrounded by a corona of epithelial cells (Fig. 1f). The epithelial corona is strongly positive for cytokeratin (Fig. 2f) but negative for vimentin (Fig. 2i). The cytokeratin is normally expressed in the tubular epithelial cells and, segmentally, in glomerular parietal epithelial cells (Fig. 2d). The rest of the glomeruli in the DDS kidneys show complex morphological changes (Fig. 1a), ranging from normal-appearing glomeruli (Fig. 1b) to gigantic proliferative (Fig. 1d) or sclerosed shrunken glomerular lesions (Fig. 1g). Intermediate spectrum of extracapillary proliferation is observed in glomeruli with or without mesangial sclerosis (Fig. 1c, e). It is worth noting that many shedding podocytes, which are strongly labeled by vimentin stain, are scattered in Bowman’s space of glomeruli with mesangial sclerosis and in tubular lumens (Fig. 2i and insert). In contrast to DDS kidneys, the non-DDS kidneys, whether harboring Wilms’ tumor (Fig. 1h) or not (Fig. 1i), show normal glomerular size without glomerular cell proliferation. Glomerular cell proliferation is demonstrated by the expression of nuclear proliferating antigens, including PCNA, Ki67 and topoisomerase IIα (TopIIα) (Fig. 3). The proliferative podocytes, which are characterized by negativity for actin (Fig. 2b) and cytokeratin (Fig. 2e) and positivity for vimentin (Fig. 2h), constantly express all proliferating antigens in hypertrophic or crescentic glomeruli in DDS kidneys (Fig. 3a, b, c, d, e, f). In contrast, endocapillary cells, which are specifically labeled by smooth-muscle actin (Fig. 2a, b, c), show little proliferative activity (Fig. 3a, b, c, d, e, f). Focal proliferation of endocapillary cells is observed only in glomeruli with advanced glomerulosclerosis (Fig. 3d, f). The assessments of cell-cycle regulatory proteins show no cyclin-A expression in either DDS or non-DDS kidneys (Fig. 4a, b). The positive cyclin-A expression in blastema cells of concurrent Wilms’ tumors serves as an internal control of cyclin-A staining (Fig. 4c). The non-DDS kidneys show extensive expression of cyclin kinase inhibitor p16 in podocytes (Fig. 5a), while DDS kidneys show loss of p16 expression in these cells (Fig. 5b). There is only mild and scattered expression of p16 in glomerular endocapillary regions of DDS kidneys (Fig. 5b). The expression of p21 (Fig. 5d) is similar to that of p16 in DDS kidneys (Fig. 5b). However, p21 expression pattern in non-DDS kidneys (Fig. 5c) differs slightly from that of p16 (Fig. 5a) in a lower labeling rate of podocytes and a higher labeling rate in parietal epithelial cells.

Light microscopy of glomeruli in Denys–Drash syndrome (DDS) kidneys (a–g) and glomeruli in non-DDS kidney (h–i). Periodic acid–Schiff stain, scale bar 50 μm

Immunohistochemical staining patterns of actin (a–c), cytokeratin (d–f) and vimentin (g–i) in Denys–Drash syndrome kidney. Peroxidase–antiperoxidase stain, scale bar 50 μm

Immunohistochemical staining patterns of cell proliferation markers: Ki67 (a, b), proliferating-cell nuclear antigen (c, d) and topoisomerase IIα (e, f) in glomeruli with early (a, c, e) and advanced (b, d, f) Denys–Drash syndrome glomerulopathy. Peroxidase–antiperoxidase stain, scale bar 50 μm

Immunohistochemical staining patterns of cyclin A in glomerulus in non-Denys–Drash syndrome (DDS) kidney (a) and in DDS kidney (b). Positive control of cyclin-A expression in concurrent Wilms’ tumor (c). Peroxidase–antiperoxidase stain, scale bar 50 μm

Immunohistochemical staining patterns of p16 (a, b) and p21 (c, d) in glomeruli in non-Denys–Drash syndrome (DDS) kidney (a, c) and in DDS kidney (b, d). Peroxidase–antiperoxidase stain, scale bar 50 μm
Assessment of WT1 expression in glomerular cells
The non-DDS kidneys constantly show diffuse global expression of WT1 on the nuclei of podocytes (Fig. 6a). The WT1 expression in the non-proliferative or mild proliferative hypertrophic glomeruli in the DDS kidneys is similar to that of non-DDS kidneys in terms of its distribution and intensity (Fig. 6b). However, the labeling intensity of WT1 gradually decreases as podocytes proliferate (Fig. 6c). WT1 expression diminishes sharply in those glomeruli with advanced extracapillary proliferation (Fig. 6d). In glomeruli with typical mesangial sclerosis, no cell of epithelial corona expresses WT1 (Fig. 6e). The glomerular endocapillary cells are constantly negative for WT1 in both DDS and non-DDS kidneys.

Immunohistochemical staining patterns of WT1 in glomerulus in non-Denys–Drash syndrome (DDS) kidney (a) and in glomeruli in DDS kidney (b–e). Peroxidase–antiperoxidase stain, scale bar 50 μm
Analysis of apoptosis in glomerular cells
The patterns of Fas ligand (Fas-L), Bax and Bcl-2 expression in the glomerular cells of the DDS kidneys and the non-DDS kidneys are depicted in Fig. 7. In the non-DDS kidneys, they are essentially absent from the cells of glomerular tufts (Fig. 7a, c, e), except for the low level of Bcl-2 expression in some glomerular parietal epithelial cells and tubular epithelial cells (Fig. 7c). Focal tubular epithelial cells also express Bax in the non-DDS kidneys (Fig. 7e). There is considerable increase of Bax (Fig. 7b) and Fas-L (Fig. 7f) expression in those glomeruli with advanced mesangial sclerosis in the DDS kidneys. The glomerular cells of both DDS and non-DDS kidneys do not express Bcl-2, except for the glomerular parietal epithelial cells (Fig. 7c, d).

Immunohistochemical staining patterns of Fas-L (a, b), Bcl-2 (c, d) and Bax (e, f) in glomeruli in non-Denys–Drash syndrome (DDS) kidney (a, c, e) and in DDS kidney (b, d, f). Peroxidase–antiperoxidase (PAP) stain, transferase-mediated dUTP nick end-labeling assay of apoptosis in glomerulus in non-DDS kidney (g) and in DDS glomerulus (h). PAP stain, scale bar 50 μm
The assessment of apoptosis using TUNEL assay reveals scattered labeling of the cells in the glomeruli that have advanced mesangial sclerosis in the DDS kidneys (Fig. 7h), whereas TUNEL-positive cells are not detected in the glomerular cells of the non-DDS kidneys (Fig. 7g). The cell origin of apoptotic cells is not readily discernible. They might be either residual podocytes or endocapillary cells. We did not observe apoptotic cells in the proliferating glomerular epithelial cells of the DDS kidneys.
Mutation analysis of WT-1
The DNA sequences of WT1 exons and exon–intron boundaries were derived from non-tumor kidney tissue of the DDS patient and the control specimens. We failed to amplify DNA of WT-1 exon 2 from the DDS patient, despite the fact that the primer set worked well on control specimens. Sequence analysis did not reveal any mutation in these DNA fragments (sequencing data not shown). Neither was there mutation in the 3′ alternative splice donor site in intron 9. There was a 915C→T (Pro→Pro) polymorphism in WT-1 exon 1 of the DDS patient.
Discussion
The WT1 expression in glomerulopathy of DDS and IDMS kidney have been reported recently by Yang et al. [33]. They demonstrated marked decrease in WT1 nuclear staining of podocytes in glomeruli that looks to have typical mesangial sclerosis [33]. The low nuclear concentration of WT1 in glomerular cells was attributed to the dominant-negative effects of mutated protein. In our study, we also observed the diminishing of WT1 in the glomerulopathy of a DDS patient. In addition, we noticed that the degree of the loss of WT1 nuclear labeling intensity was in reverse to the degree of proliferative activity of podocytes, and once glomeruli progressed to advanced glomerulosclerosis, nearly all WT1 reactivity in glomerular epithelial cells disappeared. The complete loss of WT-1 protein in DDS glomeruli with typical mesangial sclerosis might reflect the complete loss (shedding) of podocytes in these glomeruli. We speculate that the initial nuclear concentrating or binding capability of wild-type WT1 protein is not hampered because the non-proliferative glomeruli in both DDS and non-DDS kidneys have similar WT1 staining intensity. However, wt1 mutation is proposed to act in a dominant-negative manner, and a minor amount of mutant WT1 can have devastating effects [24]. Yet, it has been speculated that the loss of WT1 function in the DDS kidney might not be directly related to the effect of mutant WT1, since it has been reported that DNA binding by wild-type WT1 protein is not inhibited by an excess of mutant WT1 protein [26, 27]. This observation implies that, in addition to the alteration of DNA-binding zinc finger domain or the pertubation of WT1 isoform balance, other detrimental genetic effects might be responsible for the DDS phenotypical expression. This speculation is also supported by the fact that identical glomerulopathy can present in patients of IDMS without the evidence of mutation in all coding sequences of WT1 gene [11, 33]. In this report, we also demonstrated a DDS patient who had characteristic phenotypical manifestations of DDS but had neither Denys–Drash nor Frasier-type mutation in the wt1 gene.
Mature podocytes are constantly positive for vimentin and WT1 but negative for cytokeratin and smooth-muscle actin [10, 21]. The total loss of vimentin/WT1-positive extracapillary cells on glomerular tufts implies the elimination of podocytes in advanced DDS glomerulopathy. The observation of free-floating vimentin-positive cells in Bowman’s space and tubular lumens further suggests that shedding of podocytes from basement membrane might account, at least partly, for the loss of podocytes from DDS glomeruli. The wrapping of conglomerated glomerular tufts by cytokeratin-positive epithelial cells forms the “corona” of DMS and implicates the ingrowth of parietal or tubular epithelial cells to glomerular tufts. Whether apoptosis is responsible for the elimination of podocytes in our case of DDS remains to be determined, since we did not observe large-scale apoptosis in the glomeruli that had visceral epithelial proliferation (pictures not shown). However, several lines of evidence show that apoptosis of glomerular cells is indeed temporally and spatially related to the WT1 expression [14, 15]. Furthermore, our studies revealed some clues implicating apoptotic regulation in DDS glomerulopathy. The expression of apoptotic regulatory factors in DDS glomerulopathy depicts an apoptosis-permissive profile with increased Fas-L/Bax expression in lack of Bcl-2 activity. Both Bax, a pro-apoptotic factor, and Bcl-2, an anti-apoptotic factor, are normally absent in the podocytes of mature glomeruli [9, 13, 31]. It has been reported that Bcl-2 is a potential target of WT1 repression and spatially and temporally reverse to the WT1 expression during podocyte differentiation in the developing kidney [3, 7, 20]. However, the lack of this reciprocal correlation in DDS glomerulopathy implicates that proliferative podocytes are resistant to the induction of Bcl-2 expression and permissive for apoptosis. Since our study detected apoptotic cells only in advanced glomerulopathy of DDS, apoptosis should play a pathogenic role in advanced mesangial sclerosis and is probably initiated through Fas–L/Fas interaction in an autocrine fashion between mesangial cells or other endocapillary cells [9].
The demonstration of exaggerated podocyte proliferation in DDS glomerulopathy would address several key questions: (1) is it a common pathway to all causes of severe glomerular injury? (2) if the podocyte proliferation is a default process of DDS glomerulopathy, how does it link to WT1 mutation? The postnatal reentry of podocytes to cell cycle is definitely a pathological process, since in normal ontogenesis of human glomeruli, only immature podocytes at S-shape bodies are positive for proliferation markers Ki67 and PCNA [1, 23]. Podocytes rapidly become quiescent after the capillary-loop stage with loss of proliferation markers [23, 29]. In the present study, the podocytes in DDS glomerulopathy, regardless of proliferative or non-proliferative glomeruli, switched their cell-cycle machinery on and diffusely expressed proliferating markers, including Ki67, PCNA and topoisomerase IIα. This unique phenomenon of DDS glomerulopathy indicates that podocyte proliferation in DDS glomerulopathy is a genetic default process rather than a final result of a common pathway of non-specific glomerular injury, in which cell proliferation markers are segmentally expressed in crescentic or cellular glomerular lesions but not in mature, non-proliferating podocytes [1]. Our results also suggest that podocyte proliferation in DDS glomerulopathy may be mediated through the downregulation of both p16 and p21, which belong to the INK4 and Cip/Kip family of cyclin kinase inhibitors, respectively, and function mainly in the G1 or G1/S phase through interactions with the cyclin D-CDK complex [8, 28]. It has been reported that WT1 is able to induce p21 and is associated with G1-phase arrest and WT1-dependent differentiation pathways in the kidney [5, 21]. In experimental glomerulonephritis, the reentry of podocytes into the cell cycle is associated with the loss of WT1 and p21 expression [12]. Our results, as well as those mentioned previously, indicate the existence of an intimate relationship between WT1 and cell-cycle regulation [5, 6, 12, 32, 33]. Finally, our results also indicate that proliferation of podocytes in DDS glomerulopathy does not exactly recapture the podocyte proliferation during glomerular ontogenesis, since there is no upregulation of cyclin A [23, 29, 30].
In conclusion, the normal development and growth of glomeruli are the results of tightly controlled proliferation, differentiation and apoptosis of glomerular cells. Based on our observations, a morphological and immunohistochemical evolving process of DDS glomerulopathy is proposed (Fig. 8).

Schematic representation of morphological and immunohistochemical evolving process of Denys–Drash syndrome glomerulopathy: normal glomerulus (A); hypertrophy of glomerulus with initial hyperplasia of podocytes (B); crescentic hyperplasia of podocytes and expansion of mesangium (C); mesangial sclerosis with tubulization of Bowman’s spaces and shedding of visceral epithelial cells (D); global glomerulosclerosis (E). The following marks in parentheses indicate staining patterns: (+) positive, (+s) segmentally positive, (+w) weakly positive, (+sw) segmentally and weakly positive, (+d) positive but decrease in reactivity, (−) negative
References
Barisoni L, Kriz W, Mundel P, D’Agati V (1999) The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 10:51–61
Borel F, Barilla KC, Hamilton TB, Iskandar M, Romaniuk PJ (1996) Effects of Denys–Drash syndrome point mutations on the DNA binding activity of the Wilms’ tumor suppressor protein WT1. Biochemistry 35:12070–12076
Chandler D, el-Naggar AK, Brisbay S, Redline RW, McDonnell TJ (1994) Apoptosis and expression of the bcl-2 proto-oncogene in the fetal and adult human kidney: evidence for the contribution of bcl-2 expression to renal carcinogenesis. Hum Pathol 25:789–796
Dressler GR, Douglass EC (1992) Pax-2 is a DNA-binding protein expressed in embryonic kidney and Wilms tumor. Proc Natl Acad Sci U S A 89:1179–1183
Englert C, Maheswaran S, Garvin AJ, Kreidberg J, Haber DA (1997) Induction of p21 by the Wilms’ tumor suppressor gene WT1. Cancer Res 57:1429–1434
Grotendorst GR, Okochi H, Hayashi N (1996) A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ 7:469–480
Grubb GR, Yun K, Williams BR, Eccles MR, Reeve AE (1994) Expression of WT1 protein in fetal kidneys and Wilms tumors. Lab Invest 71:472–479
Gu Y, Turck CW, Morgan DO (1993) Inhibition of CDK2 activity in vivo by an associated 20 K regulatory subunit. Nature 366:707–710
Hattori T, Shindo S, Kawamura H (1998) Apoptosis and expression of Bax protein and Fas antigen in glomeruli of a remnant-kidney model. Nephron 79:186–191
Holthofer H, Miettinen A, Lehto VP, Lehtonen E, Virtanen I (1984) Expression of vimentin and cytokeratin types of intermediate filament proteins in developing and adult human kidneys. Lab Invest 50:552–559
Jeanpierre C, Denamur E, Henry I, Cabanis MO, Luce S, Cecille A, Elion J, Peuchmaur M, Loirat C, Niaudet P, Gubler MC, Junien C (1998) Identification of constitutional WT1 mutations, in patients with isolated diffuse mesangial sclerosis, and analysis of genotype/phenotype correlations by use of a computerized mutation database. Am J Hum Genet 62:824–833
Kim YG, Alpers CE, Brugarolas J, Johnson RJ, Couser WG, Shankland SJ (1999) The cyclin kinase inhibitor p21CIP1/WAF1 limits glomerular epithelial cell proliferation in experimental glomerulonephritis. Kidney Int 55:2349–2361
Krajewski S, Krajewska M, Shabaik A, Miyashita T, Wang HG, Reed JC (1994) Immunohistochemical determination of in vivo distribution of Bax, a dominant inhibitor of Bcl-2. Am J Pathol 145:1323–1336
Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R (1993) WT1 is required for early kidney development. Cell 74:679–691
Lipschutz JH (1998) Molecular development of the kidney: a review of the results of gene disruption studies. Am J Kidney Dis 31:383–397
Little M, Wells C (1997) A clinical overview of WT1 gene mutations. Hum Mutation 9:209–225
Little MH, Williamson KA, Mannens M, Kelsey A, Gosden C, Hastie ND, van Heyningen V (1993) Evidence that WT1 mutations in Denys–Drash syndrome patients may act in a dominant-negative fashion. Hum Mol Genet 2:259–264
Martinerie C, Chevalier G, Rauscher FJ 3rd, Perbal B (1996) Regulation of nov by WT1: a potential role for nov in nephrogenesis. Oncogene 12:1479–1492
Mueller RF (1994) The Denys–Drash syndrome. J Med Genet 31:471–477
Mugrauer G, Ekblom P (1991) Contrasting expression patterns of three members of the myc family of protooncogenes in the developing and adult mouse kidney. J Cell Biol 112:13–25
Mundel P, Reiser J, Kriz W (1997) Induction of differentiation in cultured rat and human podocytes. J Am Soc Nephrol 8:697–705
Mundlos S, Pelletier J, Darveau A, Bachmann M, Winterpacht A, Zabel B (1993) Nuclear localization of the protein encoded by the Wilms’ tumor gene WT1 in embryonic and adult tissues. Development 119:1329–1341
Nagata M, Nakayama K, Terada Y, Hoshi S, Watanabe T (1998) Cell cycle regulation and differentiation in the human podocyte lineage. Am J Pathol 153:1511–1520
Patek CE, Little MH, Fleming S, Miles C, Charlieu JP, Clarke AR, Miyagawa K, Christie S, Doig J, Harrison DJ, Porteous DJ, Brookes AJ, Hooper ML, Hastie ND (1999) A zinc finger truncation of murine WT1 results in the characteristic urogenital abnormalities of Denys–Drash syndrome. Proc Natl Acad Sci U S A 16:2931–2936
Ryan G, Steele-Perkins V, Morris JF, Rauscher FJ 3rd, Dressler GR (1995) Repression of Pax-2 by WT1 during normal kidney development. Development 121:867–875
Scharnhorst V, Dekker P, van der Eb AJ, Jochemsen AG (1999) Internal translation initiation generates novel WT1 protein isoforms with distinct biological properties. J Biol Chem 274:23456–23462
Scharnhorst V, van der Eb AJ, Jochemsen AG (2001) WT1 proteins: functions in growth and differentiation. Gene 8:141–161
Serrano M, Hannon GJ, Beach D (1993) A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366:704–707
Shankland SJ, Al’Douahji M (1999) Cell cycle regulatory proteins in glomerular disease. Exp Nephrol 7:207–211
Shankland SJ, Floege J, Thomas SE, Nangaku M, Hugo C, Pippin J, Henne K, Hockenberry DM, Johnson RJ, Couser WG (1997) Cyclin kinase inhibitors are increased during experimental membranous nephropathy: potential role in limiting glomerular epithelial cell proliferation in vivo. Kidney Int 52:404–413
Takemura T, Murakami K, Miyazato H, Yagi K, Yoshioka K (1995) Expression of Fas antigen and Bcl-2 in human glomerulonephritis. Kidney Int 48:1886–1892
Yang AH, Chen JY, Lin YP, Huang TP, Wu CW (1977) Peritoneal dialysis solution induces apoptosis of mesothelial cells. Kidney Int 51:1280–1288
Yang Y, Jeanpierre C, Dressler GR, Lacoste M, Niaudet P, Gubler MC (1999) WT1 and PAX-2 podocyte expression in Denys–Drash syndrome and isolated diffuse mesangial sclerosis. Am J Pathol 154:181–192
Acknowledgements
The present study was supported by Taipei Veterans General Hospital, under research grant no. VGH-89-140 and also by the National Science Council, ROC, under grant no. NSC 89–2320-B-075–034.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yang, A.H., Chen, J.Y. & Chen, B.F. The dysregulated glomerular cell growth in Denys–Drash syndrome. Virchows Arch 445, 305–314 (2004). https://doi.org/10.1007/s00428-004-1069-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-004-1069-2