
Abstract
We identified 5′ upstream enhancers of two Ci-ZicL genes and characterized one of them in detail. Although the genes are tandemly repeated in the genome, the transcription of each seemed to be individually regulated. The 259-bp 5′ flanking sequence contained essential elements for driving a correct spatiotemporal expression. This enhancer can be divided into two distinct modules. The A module was located between nucleotide positions −259 and −205 upstream of the putative transcription start site, and was necessary for activation in A6.2 and A6.4 blastomeres at the 32-cell stage. The BM module lay between nucleotide positions −205 and −89 and was responsible for activation in B6.2 and B6.4 blastomeres at the 32-cell stage and in A-line presumptive notochord, nerve cord, and muscle lineage cells at later stages. Two putative Fox-binding sites, one located within and the other downstream of the BM module, were necessary for the latter activity. Mutation at a potential Ets-binding site, located downstream of the BM module, caused ectopic activation of the reporter gene in a-line presumptive ectoderm cells. This suggests that repression in the a-line blastomeres is necessary for correct transcriptional control of the Ci-ZicL gene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the goals of developmental biology is to elucidate the complete genetic cascade from fertilized egg to terminally differentiated tissues. Protochordate ascidians offer advantages in studying this issue, as the fate of most embryonic cells is determined during the first four to 5 h of embryogenesis (for review, see Nishida 2005). The pattern of cleavage in early ascidian embryos is invariant, and the cell lineage has been precisely described (Conklin 1905; Nishida 1987). The notochord is the tissue after which “chordates” are named. Its pathway of differentiation in ascidian embryos has been well-documented in three species, Ciona intestinalis, Ciona savignyi, and Halocynthia roretzi (for reviews, see Di Gregorio and Levine 1998; Nishida 2005; Satoh 2003; Satou and Satoh 1999).
Brachyury, a master regulatory gene, is expressed in the notochord lineage cells from the 64-cell stage (Yasuo and Satoh 1993; Corbo et al. 1997b). Brachyury target genes responsible for the function of the notochord were characterized (Takahashi et al. 1999; Hotta et al. 2000). Brachyury-responsive enhancer activity was demonstrated for one of them (Di Gregorio and Levine 1999). The expression of Brachyury and differentiation of the notochord are induced by vegetally located presumptive endoderm blastomeres at the 32-cell stage (Nakatani and Nishida 1994; Nakatani et al. 1996). The inductive signal is thought to be the basic fibroblast growth factor (bFGF) (Nakatani et al. 1996; Kim and Nishida 2001). The bFGF gene, Fgf9/16/20 (originally named Fgf4/6/9), is expressed downstream of the β-catenin gene in vegetal blastomeres at the 16-cell and 32-cell stages (Imai et al. 2002a).
However, the notochord specification is not simple. An Fgf9/16/20-specific morpholino oligo did not completely block the expression of Brachyury (Imai et al. 2002a). Expression of Brachyury is also activated by a zinc finger-containing transcription factor, ZicL (Imai et al. 2002c; Wada and Saiga 2002; Yagi et al. 2004). Expression of HrzicN, a Halocynthia ZicL homolog, was not dependent on a bFGF-like signal (Wada and Saiga 2002). These observations suggest that the specification of the notochord is regulated by two or more relatively independent pathways (Imai et al. 2002c; Wada and Saiga 2002).
ZicL (Ci-ZicL) was identified as an expressed sequence tag (EST) (cluster ID 03695r1), a blastomere-specific zygotic expression of which was observed in the cleavage-stage C. intestinalis embryo (Fujiwara et al. 2002). ZicL (Cs-ZicL) had been independently isolated as a β-catenin downstream target in C. savignyi (Imai et al. 2002c). The ZicL gene and its orthologs are expressed in the equatorial region (A6.2, A6.4, B6.2, and B6.4 blastomere pairs) of the 32-cell embryo (Imai et al. 2002c; Wada and Saiga 2002; Yagi et al. 2004; see Fig. 1). These blastomeres include the notochord lineage.

The 5′ flanking regions of Ci-ZicL genes. a Two different 5′ flanking sequences of Ci-ZicL genes (Ci-ZicL-A and Ci-ZicL-B) were isolated, which seemed to correspond to tandemly repeated genes in the Ciona genome. Restriction sites are indicated. b The alignment of two upstream regions. Putative binding sequences for the Ets-domain transcription factor (Ets) and Fox proteins (Fox) are boxed. The binding sites in the Ci-ZicL-B sequence are numbered. Restriction sites are indicated. A single E-box was found, and this is a PmlI-recognition site. The transcription start site was deduced from the longest cDNA clone (data not shown)
The next step in tracking the genetic cascade of notochord specification back to the fertilized egg is to reveal the regulatory mechanism of the blastomere-specific expression of the ZicL gene. Transcription of Cs-ZicL in A6.2 and A6.4 blastomeres seemed to be activated by the winged-helix transcription factor Cs-FoxD in C. savignyi, as suppression of the function of Cs-FoxD with a specific morpholino oligo abolished the expression of Cs-ZicL in these blastomeres (Imai et al. 2002c). The expression of Cs-FoxD is directly activated by the nuclear accumulation of β-catenin (Imai et al. 2002b). This is a clue to unraveling the ZicL transactivation mechanism in the early ascidian embryo, although it is unclear whether the activation is direct.
In the present study, upstream sequences of two Ci-ZicL genes were isolated from the genomic DNA of C. intestinalis. The enhancer module required for the expression in A6.2 and A6.4 blastomeres can be separated from that required for the expression in B6.2 and B6.4 blastomeres. After the next cell division, the latter enhancer module was activated in all the daughter cells of A6.2, A6.4, B6.2, and B6.4 blastomeres. Putative Fox-binding sites seemed important for the activation of the enhancer in A-line blastomeres. A binding site for Ets-domain transcription factors was necessary for repression of the enhancer in the ectodermal lineage blastomeres in the animal hemisphere. The expression of Ci-ZicL was intricately regulated in a blastomere-specific manner.
Materials and methods
Animals
Adult C. intestinalis was collected or cultured using plastic seeding pots in the Uranouchi Inlet near the Usa Marine Biological Institute of Kochi University. The animals were also kindly provided by Kazuko Hirayama and Nori Satoh of Kyoto University, Takahito Nishikata of Konan University, and Hiroki Takahashi of the National Institute for Basic Biology. Adults were maintained in a natural seawater aquarium at 16°C under constant light. Gametes were surgically obtained from the gonoducts. Eggs were inseminated with non-self sperm and dechorionated with 0.05% actinase E (Kaken) and 1% sodium thioglycolate. Embryos were raised at 18°C.
Isolation of Ci-ZicL 5′ flanking regions
The regions upstream of two Ci-ZicL genes (named Ci-ZicL-A and Ci-ZicL-B) were amplified by PCR. The primers used to amplify Ci-ZicL-A were 5′-GTTTATTAGGTCAGTTTC-3′ and 5′-ACTTGCAAGCAATCGGAT-3′. Those used to amplify Ci-ZicL-B were 5′-TGCAATTGCGTATCTTGT-3′ and 5′-ACTTGCAAGCAATCGGAT-3′. The PCR products were subcloned into pGEM-T (Promega). Nucleotide sequences were determined using a BigDye Terminator v3.1 cycle sequencing kit and PRISM 3100-Avant Genetic Analyzer (Applied Biosystems).
Construction of plasmids
The Ci-ZicL-B insert was amplified from pGEM-T-Ci-ZicL-B by PCR, using primers 5′-AGCAAGCTTTTCTTGTTTTTATTAAATTTGC-3′ and 5′-AACTGCAGCAACCATTACATTAGAATA-3′. The PCR product was digested with HindIII and PstI and replaced with the Ci-Bra enhancer of the Ci-Bra-lacZ plasmid construct (Corbo et al. 1997b). The resultant plasmid, named Ci-ZicL-B/lacZ, was used to produce the following constructs. To generate −658Z, −295Z, −259Z, −205Z, −127Z and −89Z, Ci-ZicL-B/lacZ was linearized with SpeI, BstBI, SspI, SnaBI, NruI, and PmlI, respectively. Distal regions of the enhancer were then excised with XhoI. The cohesive ends were blunted using T4 DNA polymerase, and circular plasmids were recovered by self-ligation. For construction of −507Z, −411Z, and −356Z, PCR was carried out using a lacZ-specific primer (5′-TAGCGCCGAGTCAAGCT-3′) and a Ci-ZicL-B-specific primer (5′-ACGCTCAAAACTCGAGATTGCGTAAATGCTACTTCAA-3′ for −507Z, 5′-AATGTCCTACTCGAGTTTTGAACAATTAACAACGG-3′ for −411Z, and 5′-TCTTAACTACCTCGAGGGAAGAGAAAGTAGAATGAAA-3′ for −356Z). The PCR products were digested with XhoI and PstI, and replaced with the region upstream of Ci-ZicL-B/lacZ. ΔBSn, ΔSnN, ΔNP, and ΔSnP constructs were obtained by cutting out the BstBI-SnaBI, SnaBI-NruI, NruI-PmlI, and SnaBI-PmlI fragments from −658Z, respectively.
Site-directed mutagenesis was performed according to the overlap extension method (Sambrook and Russell 2001). The Ci-ZicL-B enhancer fragment was excised from pGEM-T-Ci-ZicL-B using XhoI and PstI, and inserted into pBluescript II SK+ (Stratagene). Overlapped mutagenic primers for a potential Ets-binding site (Ets4) were 5′-AAGCGACGGGCGATCAAGAGTCAAATAAATAAA-3′ and 5′-TTTATTTATTTGACTCTTGATCGCCCCGTCGTCT-3′. Mutagenic primer sets for putative Fox-binding sites (Fox2, Fox4 and Fox5) were (5′-CGAAAAACGAATTGGGCGTGTTTTACTTGTGT-3′ and 5′-ACACAAGTAAAACACGCCCAATTCGTTTTTCG-3′), (5′-TTACCTTCGCGAGGCGGGTATCGGAGGTGGGGG-3′ and 5′-CCCCCACCTCCGATACCCGCCTCGCGAAGGTAA-3′) and (5′-GATCGGAAGTCAAATGGGTAAAGATAAAGAAGA-3′ and 5′-TCTTCTTTATCTTTACCCATTTGACTTCCGATC-3′), respectively. Mutagenized enhancers were obtained by PCR using M13-forward, M13-reverse, and the above primer sets. The PCR products were first digested with SpeI or SnaBI (blunted with T4 DNA polymerase), then cut with PstI and replaced with the enhancer fragment of Ci-ZicL-B/lacZ.
Electroporation and in situ hybridization
Large-scale preparation of plasmid DNA was carried out using QIAGEN tip-100 (Qiagen). The introduction of reporter genes into dechorionated Ciona embryos by electroporation was performed as described by Corbo et al. (1997b). Some embryos were treated from the 24-cell stage with 2 μM of the MEK inhibitor U0126 (Promega), as described by Kim and Nishida (2001). Reporter gene expression was examined by in situ hybridization using a lacZ-specific RNA probe, essentially according to the protocol described by Satou et al. (1995).
For the in situ detection of Ci-ZicL and Ci-FoxD mRNAs, cDNA clones (ID 03695r1 and 01293r1, respectively) from EST libraries were used as a probe (Fujiwara et al. 2002; Satou et al. 2002). In situ hybridization using DNA probes was carried out according to Fujiwara et al. (2002).
Results
Characterization of the Ci-ZicL 5′ upstream sequences
Five Ci-ZicL genes (Ci-ZicL1~5) were identified in the Ciona intestinalis genome (Yamada et al. 2003). The nucleotide identity of any two of them was about 90%. Their predicted amino acid sequences were almost identical. To identify cis-regulatory elements of Ci-ZicL genes, we isolated two different genomic DNA fragments. Each of them contained a Ci-ZicL promoter region, including putative transcription and translation initiation sites (Fig. 1). Their nucleotide sequences seemed to correspond to tandemly repeated Ci-ZicL genes (Fig. 1a). However, we could not conclude which of the five genes were contained in our DNA fragments due to the presence of two sets of very similar tandem repeats (Yamada et al. 2003) and the high polymorphism of the Ciona genome (Dehal et al. 2002). We, therefore, call the two genes Ci-ZicL-A and Ci-ZicL-B for convenience (Fig. 1a). Their nucleotide sequences were 99% identical within the proximal 240 bp of the 5′ flanking region (Fig. 1b). This region contained an E-box (CANNTG), a potential binding site for Ets-domain transcription factors [NNA(A/C)(A/C)GGA(A/T)(A/G)(A/T)NN], and two putative binding sites for winged-helix/Fox transcription factors [NNNT(A/G)TTT(A/G)(C/T)T(C/T)] (Fig. 1b). Distal sequences were not conserved (Fig. 1b).
An enhancer sufficient for correct transcriptional regulation
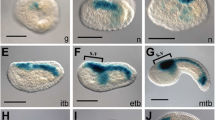
We tested whether each DNA fragment contained all the information necessary for a correct spatiotemporal expression. For this purpose, we first examined the precise expression pattern of the endogenous Ci-ZicL gene in the early Ciona embryo (Fig. 2a–f). Ci-ZicL mRNA was first detected in A6.2, A6.4, B6.2, and B6.4 blastomere pairs (Fig. 2a; Yagi et al. 2004). After the next cleavage, all of the daughter cells expressed the mRNA (Fig. 2b). Among them A7.3, A7.7, B7.3, and B7.7 give rise to the notochord and mesenchyme, depending on the induction. The other daughter cells (A7.4, A7.8, B7.4, and B7.8) give rise to the nerve cord and muscle. We found that Ci-ZicL expression became transiently strong in the presumptive notochord/mesenchyme cells around the 76-cell stage (Fig. 2c). At the 110-cell stage, the amount of Ci-ZicL mRNA in these cells decreased, and instead, that in the nerve cord/muscle lineage cells transiently increased (Fig. 2d,e). From this stage, presumptive brain/nerve cord cells derived from the anterior animal (a-line) blastomeres started to express Ci-ZicL mRNA (Fig. 2e,f). At the 44-cell stage, B6.3 started to express Ci-ZicL (Fig. 2b). One of its daughter cells, B7.5, strongly expressed Ci-ZicL (Fig. 2c–f). Dynamic changes in the expression pattern suggest that distinct regulatory mechanisms drive Ci-ZicL transcription in different lineages.

The 658 bp of the 5′ flanking region of Ci-ZicL-B drives lacZ reporter gene expression with the correct spatiotemporal pattern. Stained embryos in all panels are oriented to display the vegetal side, with the anterior side up. Since the cleavage pattern is bilaterally symmetrical, stained blastomeres are indicated only on the right side. (a–f) The expression pattern of the endogenous Ci-ZicL gene. Presumptive fates of the stained cells are indicated in (c, d). Me mesenchyme, Mu muscle, Nc nerve cord, No notochord. Green arrowheads show the a-line presumptive CNS cells. (g–i) Expression pattern of the lacZ reporter gene containing the 658 bp of the Ci-ZicL-B 5′ flanking region. Expression of the reporter gene was detected by in situ hybridization
The Ci-ZicL-A and Ci-ZicL-B genomic DNA fragments were fused in-frame to lacZ and introduced into the one-cell embryo by electroporation. The transgene, named −658Z, containing 658 bp of the Ci-ZicL-B sequence immediately upstream of the putative transcription start site, was expressed in A6.2, A6.4, B6.2, and B6.4 at the 32-cell stage (Fig. 2g). The daughter cells expressed this transgene in the 44-cell embryo (Fig. 2h). At the 110-cell stage, expression of −658Z was detected in those cells that normally expressed endogenous Ci-ZicL (Fig. 2i, compare with Fig. 2d). A delicate spatiotemporal difference in the amount of lacZ transcript was not detected. However, this was probably because lacZ mRNA was relatively stable, and because the transgene was unevenly distributed between blastomeres. The reporter gene fused to the 0.7-kb Ci-ZicL-A upstream region gave an identical pattern of expression (data not shown). The Ci-ZicL-A and Ci-ZicL-B sequences used for this analysis did not overlap (Fig. 1a), suggesting that the transcription of each of the tandemly repeated Ci-ZicL genes is independently controlled. The Ci-ZicL-binding sequence (Yagi et al. 2004) was not found. This suggests that the 658-bp enhancer did not contain an autoregulatory maintenance element (see Fig. 1b). We used the Ci-ZicL-B enhancer for further analysis.
An enhancer module required for activation in A6.2 and A6.4 at the 32-cell stage
We carried out a series of deletion analyses (Fig. 3). Transgenes containing 658, 507, 411, 356, or 295 bp of the 5′ flanking sequence gave similar expression patterns at the 32-cell stage, although the enhancer activity gradually decreased as the enhancer was shortened (Fig. 3b,c; and data not shown). The transgene containing 205 bp of the 5′ flanking region, named −205Z, was expressed in B6.2 and B6.4, but not in A6.2 and A6.4 (Fig. 3e). We generated an internal deletion of the region between nucleotide positions −295 and −205 upstream of the transcription start site of the −658Z transgene (named ΔBSn; see Fig. 3a). Expression of ΔBSn was activated only in B6.2 and B6.4 (Fig. 3f). The deleted region was characterized by two potential binding sites for Fox transcription factors (designated Fox2 and Fox3; Figs. 1a and 3a). The −259Z transgene did not contain Fox2 and Fox3 (Fig. 3a), but was expressed in A6.2 and A6.4 (Fig. 3d). Similarly, mutation of the Fox2 sequence did not abolish lacZ expression in A6.2 and A6.4 (Fig. 3g). These results suggest that the region between nucleotide positions −259 and −205, named the “A module” is essential for activation in A6.2 and A6.4. Additional Fox-binding sites (named Fox4 and Fox5) were found downstream of the A module (Figs. 1a and 3a). Simultaneous mutations of the Fox2, Fox4, and Fox5 sequences diminished lacZ expression in A6.2 and A6.4, but not in B6.2 and B6.4 (Fig. 3h). The expression was not completely abolished even in A-line cells (Fig. 3h, arrowheads). Expression of Ci-FoxD in the C. intestinalis embryo was examined by in situ hybridization. The Ci-FoxD mRNA was detected in A5.1, A5.2, and B5.1 blastomeres of the 16-cell embryo (Fig. 3i), as was observed for Cs-FoxD in the C. savignyi embryo (Imai et al. 2002b).

Identification of an enhancer module necessary for activation in the 32-cell embryo. a The diagrams show the different 5′ flanking regions used for the analysis in b–h. Names of the transgenes are on the right side. Four putative Fox-binding sites (Fox2~5) are indicated by boxes. b–h The expression pattern of transgenes, −658Z, −295Z, −259Z, −205Z, ΔBSn, Fox2-mut, and Fox2/4/5-mut. All panels show the vegetal side of the 32-cell embryos. The anterior side is up. Stained blastomeres are indicated. Arrowheads in (h) show the expression of the Fox2/4/5-mut transgene in A6.2 and A6.4. i The expression pattern of Ci-FoxD mRNA in the 16-cell embryo. The embryo is oriented to show the vegetal side, with the anterior side up. j Different 5′ flanking regions that were used for the analysis shown in k–n. k–n The expression pattern of transgenes, −127Z, ΔSnN, ΔNP, ΔSnP, and −205Z, at the 32-cell stage. All panels show the vegetal side, with the anterior side up. Stained blastomeres are indicated
An enhancer module required for activation in B6.2 and B6.4 at the 32-cell stage
As the −205Z transgene seemed to include sequence motifs sufficient for the expression in B6.2 and B6.4 (Fig. 3e), we carried out further deletion analyses (Fig. 3j). The transgene containing 127 bp of the 5′ flanking region was very weakly activated, and in many cases the expression was observed only in B6.4 (Fig. 3k). No expression was obtained with the transgene containing the 89-bp upstream sequence (data not shown). As further deletion may cause disruption of the basal promoter, we generated an internal deletion of the region between nucleotide positions −205 and −127, −127, and −89, or −205 and −89 of the −658Z transgene (Fig. 3j). These transgenes were named ΔSnN, ΔNP, and ΔSnP, respectively (Fig. 3j). Expression of ΔSnN was weak but detected not only in A6.2 and A6.4 but also in B6.2 and B6.4 (Fig. 3l). ΔNP gave a strong expression comparable to −658Z (Fig. 3m; see also Figs. 2g and 3b). In contrast, expression of ΔSnP was faint in B6.4 and not observed in B6.2 (Fig. 3n). These results suggest that the region between nucleotide positions −205 and −89 is an enhancer module necessary for activation in B6.2 and B6.4. We call this region the BM module (see Fig. 5). A single potential Fox-binding site (Fox4) and E-box were located in this module (Fig. 1b). However, disruption of either one of these sites on −658Z did not alter the expression pattern in the 32-cell embryo (data not shown). Another Fox-binding site (Fox5) was found within the 89-bp upstream sequence, outside of the BM module. However, disruption of this site did not have any effect either (data not shown).
The BM module is required for activation in A-line presumptive notochord, nerve cord, and muscle cells at later stages
We showed that −658Z was expressed in the daughter cells of A6.2, A6.4, B6.2, and B6.4 (Fig. 2h). Among the daughter cells, A7.3, A7.7, B7.3, and B7.7 give rise to the notochord and mesenchyme by induction. Ci-ZicL expression was transiently upregulated in these cells (Fig. 2d). This suggested the existence of a distinct mechanism driving Ci-ZicL expression in the notochord/mesenchyme lineage cells.
We found that the −205Z transgene was expressed in all the daughter cells of A6.2, A6.4, B6.2, and B6.4 (Fig. 4b,c). Note that the A module, essential for the activation of transgenes in A6.2 and A6.4, was not included in −205Z (Figs. 3 and 5). Actually, −205Z was not expressed in A6.2 and A6.4 at the 32-cell stage (Fig. 3e). These results suggest that the lacZ mRNA detected in A7.3, A7.4, A7.7, and A7.8 was not inherited from the mother cells (A6.2 and A6.4) but was newly expressed in these cells by a distinct mechanism. At present, however, it is not clear whether the mechanism of activation in B7.3, B7.4, B7.7, and B7.8 is different from that in their mother cells (B6.2 and B6.4). Deletion of the BM module from −205Z resulted in a complete loss of expression (data not shown), suggesting that this module contains elements necessary for activation in A-line cells after the 44-cell stage.

a–d Activation of the enhancer in A-line blastomeres at later stages. a The transgenes that were used for the analysis shown in b–d. Two putative Fox-binding sites (Fox4 and Fox5) are indicated by boxes. These sites were disrupted in Fox4/5-mut. b Expression of −205Z in the 44-cell embryo (compare with Fig. 2h). c Expression of −205Z in the early gastrula. Me mesenchyme, Mu muscle, Nc nerve cord, No notochord. d Expression of Fox4/5-mut in the 64-cell embryo. All panels show the vegetal view, with the anterior side up. e–g MEK-dependent repression of the enhancer in the ectodermal lineage. e The transgenes that were used for the analysis shown in f and g. An Ets-binding site (Ets4) in −658Z is indicated by a box. This site was disrupted in Ets-mut. f Ectopic expression of Ets-mut in the a-line blastomeres of the 32-cell embryo. g Ectopic expression of −658Z in the a-line blastomeres of the 32-cell embryo, treated with the MEK inhibitor, U0126 (compare with Fig. 3b)

Summary of the cis-regulatory elements in the Ci-ZicL-B enhancer. The enhancer contains two modules, the A module and BM module. The former is necessary for activation in A6.2 and A6.4, while the latter is required for activation in B6.2 and B6.4 at the 32-cell stage. Putative Fox-binding sites (Fox2~5) and an Ets-binding site (Ets4) are indicated by boxes. At later stages, the BM module is required for activation in A-line presumptive notochord, nerve cord, and muscle cells. The Fox4 and Fox5 sequences are necessary for this activity. The Ets4 site is required for repression in a-line ectodermal lineage blastomeres
Mutations were generated at two putative Fox-binding sites (Fox4 and Fox5) on the −205Z transgene (Fig. 4a). The mutant transgene, named Fox4/5-mut, was expressed only in B-line cells (Fig. 4d). The result suggests that Fox transcription factor(s) are necessary for the activation of the BM module in A-line cells but not in B-line cells.
The Ci-ZicL enhancer activity is repressed in a-line presumptive ectoderm blastomeres
The notochord and mesenchyme in ascidians differentiate in response to bFGF-like signals (Nakatani et al. 1996; Kim and Nishida 2001), and the signals are mediated by an Ets-domain transcription factor (Miya and Nishida 2003). The Ci-ZicL-B enhancer contained four putative binding sites for Ets-domain proteins (Fig. 1b). As the endogenous Ci-ZicL gene was transiently upregulated and −205Z was expressed in the notochord/mesenchyme lineage cells, we tested the importance of one of the four binding sites (named Ets4) that was included in the 205-bp enhancer (Fig. 4e). Conversion of Ets4 (CGATCGGAAGTCA) into (CGATCAAGAGTCA) did not diminish the expression in the notochord/mesenchyme lineage, but resulted in an unexpected ectopic activation in the a-line presumptive ectoderm blastomeres (Fig. 4f). The ectopic expression was observed in a6.5, a6.6, a6.7, and a6.8, which give rise to the epidermis and central nervous system (CNS). In the early gastrula, Ci-ZicL mRNA was expressed in the a-line CNS precursor, but not in prospective epidermal cells (Fig. 2e,f; Yagi et al. 2004). Even in the CNS lineage cells, Ci-ZicL expression was not detected at the 32-cell stage (Fig. 2). To examine whether the ectopic activation depends on MEK/MAPK signal transduction, expression of the −658Z transgene was analyzed in the embryos treated with the MEK inhibitor, U0126. Ectopic expression was observed in the ectodermal lineage blastomeres of the treated embryos (Fig. 4g). Control (dimethylsulfoxide-treated) embryos did not show ectopic expression (data not shown). The expression in the notochord/mesenchyme lineage was not affected by U0126 treatment (Fig. 4g). These results suggest that, in a-line blastomeres, a MEK/MAPK-mediated extracellular signal represses Ci-ZicL transcription via an Ets-domain transcription factor. The expression in the notochord/mesenchyme lineage does not seem to require MEK/MAPK/Ets signal transduction.
Discussion
In the present study, we characterized the 5′ upstream enhancer of one of the five Ci-ZicL genes (Ci-ZicL-B). A series of deletion analyses revealed two distinct enhancer modules (Fig. 5). The A module is the region between nucleotide positions −259 and −205 upstream of the putative transcription start site (Fig. 5). This module was necessary for activation in A6.2 and A6.4 at the 32-cell stage. The BM module lay between nucleotide positions −205 and −89, and was required for activation in B6.2 and B6.4 at the 32-cell stage and in A-line presumptive notochord, nerve cord, and muscle cells at later stages (Fig. 5). The ectopic activation of the reporter gene in a-line presumptive ectoderm blastomeres, caused by a mutation in a potential Ets-binding site, suggests that repression in the a-line cells is necessary for the correct transcriptional control of the Ci-ZicL gene. Possible regulatory mechanisms are discussed below.
The activation of the A module
The A module does not contain putative Fox-binding site. This is inconsistent with the fact that a FoxD-specific morpholino oligo inhibited the expression of the ZicL gene in A6.2 and A6.4 of the 32-cell C. savignyi embryo (Imai et al. 2002c). In vitro translated Ci-FoxD and Ci-FoxA proteins did not show DNA-binding activity (data not shown). Therefore, we cannot conclude whether Fox proteins bind to the putative binding sites or elsewhere in the Ci-ZicL enhancer. It is also unclear whether the possible binding of Fox proteins is inhibited by the mutagenesis carried out in this study. Further investigation is necessary to determine whether FoxD binds and directly activates the A module.
The activation of the BM module
The BM module is required for activation in B6.2 and B6.4 at the 32-cell stage, and in A7.3, A7.4, A7.7, and A7.8 at later stages. As for the former activity, we could not find a potential transcription-factor-binding site that was necessary for the activation of the BM module in B6.2 and B6.4. Site-specific mutagenesis at putative Fox-binding sites and in an E-box did not diminish the expression. Yagi et al. (2005) showed that knockdown of both Ci-Tbx6b and Ci-Tbx6c resulted in the suppression of Ci-ZicL expression in B6.2. The expression pattern of Ci-ZicL in the double knockdown embryos looks similar to that of the ΔSnP reporter gene in the present study (Yagi et al. 2005), although we did not find potential Ci-Tbx6-binding sites within the BM module.
Activation of the Ci-ZicL enhancer in the notochord/mesenchyme lineage cells seemed independent of the MEK/MAPK signal transduction pathway. This coincides with the result that Hr-ZicN mRNA was detected in MEK-inhibitor-treated Halocynthia embryos (Wada and Saiga 2002). Two Fox-binding sites (Fox4 and Fox5) were necessary for the activation of the BM module in A-line presumptive notochord, nerve cord, and muscle cells after the 44-cell stage. Ci-FoxD and Ci-FoxA are candidate transcriptional activators. Ci-FoxD mRNA was detected in A5.1 and A5.2 of the 16-cell embryo. A5.1 divides into A6.1 and A6.2, while A5.2 divides into A6.3 and A6.4. However, Ci-ZicL is expressed only in A6.2 and A6.4, not in A6.1 and A6.3. One possible mechanism is an uneven distribution of Ci-FoxD protein into one of the daughter cells. Another possibility is that the Ci-FoxD protein exists in all the descendant cells of A5.1 and A5.2, and the Ci-ZicL is repressed by another factor in A6.1 and A6.3. Ci-FoxA, formerly called Ci-fkh (Corbo et al. 1997a), is expressed in A5.1, A5.2 and B5.1 of the 16-cell embryo, and most of their daughter cells including A6.2 and A6.4, of the 32-cell embryo (Di Gregorio et al. 2001). Also in this case, the question why Ci-ZicL is not expressed in all of the Ci-FoxA-expressing cells remains to be answered.
Repression of Ci-ZicL expression in the ectodermal lineage
A MEK-mediated signal transduction and Ets-domain transcription factor(s) seemed to be involved in the repression of Ci-ZicL in the a-line ectodermal blastomeres. A simple explanation for this is as follows. An Ets-domain transcriptional repressor is expressed in the a-line blastomeres. The repressor activity is stimulated by an extracellular signal mediated by MEK. An Ets-domain protein, ESE-3, functions as a MEK/MAPK-dependent transcriptional repressor in human cell lines (Tugores et al. 2001). A Ciona homolog of ESE (ID 30129r1) was identified by EST and genome analyses (Nishikata et al. 2001; Yagi et al. 2003). The ESE cDNA was obtained from the one-cell embryo, suggesting that ESE protein is maternally provided, although its localization has not been examined. FGF9/16/20 induces the expression of Ci-otx in a6.5 of the 32-cell embryo, dependent on MEK and Ets (Bertrand et al. 2003). Therefore, a-line blastomeres are fully equipped with the essential components of the MEK/MAPK/Ets signal transduction pathway. The human ESE is thought to function as a context-dependent (promoter-dependent) transcriptional activator or repressor (Silverman et al. 2002). A Ciona Ets-domain factor may activate Ci-otx expression and repress Ci-ZicL expression in a6.5. However, Ci-otx is not expressed in a6.6, a6.7, and a6.8, where the ectopic activation of Ci-ZicL transgenes was observed. Questions remain which cannot be easily answered.
A MAPK-dependent Ets-domain transcriptional activator, Elk-1, seems to act as a repressor without stimulation by MAPK in the mammalian system (Yang et al. 2002). This raises another possibility. An Ets-domain factor binds to the Ci-ZicL enhancer and represses transcription in the a-line blastomeres without stimulation by the MEK/MAPK signal transduction pathway. This or another Ets-domain factor then activates transcription in the CNS upon stimulation by FGF (Lemaire et al. 2002; Hudson et al. 2003; Bertrand et al. 2003). The third possibility is that the mutagenesis at the Ets-binding site unexpectedly created a binding site for another transcriptional activator. However, the latter two hypotheses can hardly explain the ectopic expression of the transgene caused by the MEK inhibitor treatment. The first possibility is the simplest and most likely, but further analysis is necessary.
Conclusions and perspectives
We characterized the enhancer modules responsible for the blastomere-specific activation of Ci-ZicL expression. Ci-ZicL seemed to be activated by several distinct transcriptional regulatory mechanisms as early as the 32-cell stage. We narrowed each essential module down to about 100 bp. This will facilitate the search for relevant transcription factors. We are now preparing an embryonic nuclear extract to identify the proteins that specifically bind to the enhancer fragments. The possible involvement of the Ets-domain transcription factor in the repression of Ci-ZicL transcription in a-line blastomeres has raised complicated questions. Mechanisms involved in this process may also be responsible for activation of Ci-ZicL in the CNS. In the present study, we focused our attention on the initiation of Ci-ZicL transcription before gastrulation. Further analysis is necessary for unraveling the mechanism of repression and activation of Ci-ZicL transcription in a-line cells.
References
Bertrand V, Hudson C, Caillol D, Popovici C, Lemaire P (2003) Neural tissue in ascidian embryos is induced by FGF9/16/20, acting via a combination of maternal GATA and Ets transcription factors. Cell 115:615–627
Conklin EG (1905) The organization and cell lineage of the ascidian egg. J Acad Nat Sci Phila 13:1–119
Corbo JC, Erives A, Di Gregorio A, Chang A, Levine M (1997a) Dorsoventral patterning of the vertebrate neural tube is conserved in a protochordate. Development 124:2335–2344
Corbo JC, Levine M, Zeller RW (1997b) Characterization of a notochord-specific enhancer from the Brachyury promoter region of the ascidian, Ciona intestinalis. Development 124:589–602
Dehal P, Satou Y, Campbell RK, Chapman J, Degnan B, De Tomaso A, Davidson B, Di Gregorio A, Gelpke M, Goodstein DM, Harafuji N, Hastings KEM, Ho I, Hotta K, Huang W, Kawashima T, Lemaire P, Martinez D, Meinertzhagen IA, Necula S, Nonaka M, Putnam N, Rash S, Saiga H, Satake M, Terry A, Yamada L, Wang H-G, Awazu S, Azumi K, Boore J, Branno M, Chin-bow S, De Santis R, Doyle S, Francino P, Keys DN, Haga S, Hayashi H, Hino K, Imai KS, Inaba K, Kano S, Kobayashi K, Kobayashi M, Lee B-I, Makabe KW, Manohar C, Matassi G, Medina M, Mochizuki Y, Mount S, Morishita T, Miura S, Nakayama A, Nishizaka S, Nomoto H, Ohta F, Oishi K, Rigoutsos I, Sano M, Sasaki A, Sasakura Y, Shoguchi E, Shin-i T, Spagnuolo A, Stainier D, Suzuki MM, Tassy O, Takatori N, Tokuoka M, Yagi K, Yoshizaki F, Wada S, Zhang C, Hyatt PD, Larimer F, Detter C, Doggett N, Glavina T, Hawkins T, Richardson P, Lucas S, Kohara Y, Levine M, Satoh N, Rokhsar DS (2002) The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science 298:2157–2167
Di Gregorio A, Corbo JC, Levine M (2001) The regulation of forkhead/HNF-3β expression in the Ciona embryo. Dev Biol 229:31–41
Di Gregorio A, Levine M (1998) Ascidian embryogenesis and the origins of the chordate body plan. Curr Opin Genet Dev 8:457–463
Di Gregorio A, Levine M (1999) Regulation of Ci-tropomyosin-like, a Brachyury target gene in the ascidian, Ciona intestinalis. Development 126:5599–5609
Fujiwara S, Maeda Y, Shin-i T, Kohara Y, Takatori N, Satou Y, Satoh N (2002) Gene expression profiles in Ciona intestinalis cleavage-stage embryos. Mech Dev 112:115–127
Hotta K, Takahashi H, Asakura T, Saitoh B, Takatori N, Satou Y, Satoh N (2000) Characterization of Brachyury-downstream notochord genes in the Ciona intestinalis embryo. Dev Biol 224:69–80
Hudson C, Darras S, Caillol D, Yasuo H, Lemaire P (2003) A conserved role for the MEK signalling pathway in neural tissue specification and posteriorization in the invertebrate chordate, the ascidian Ciona intestinalis. Development 130:147–159
Imai KS, Satoh N, Satou Y (2002a) Early embryonic expression of FGF4/6/9 gene and its role in the induction of mesenchyme and notochord in Ciona savignyi embryos. Development 129:1729–1738
Imai KS, Satoh N, Satou Y (2002b) An essential role of a FoxD gene in notochord induction in Ciona embryos. Development 129:3441–3453
Imai KS, Satou Y, Satoh N (2002c) Multiple functions of a Zic-like gene in the differentiation of notochord, central nervous system and muscle in Ciona savignyi embryos. Development 129:2723–2732
Kim GJ, Nishida H (2001) Role of the FGF and MEK signaling pathway in the ascidian embryo. Dev Growth Differ 43:521–533
Lemaire P, Bertrand V, Hudson C (2002) Early steps in the formation of neural tissue in ascidian embryos. Dev Biol 252:151–169
Miya T, Nishida H (2003) An Ets transcription factor, HrEts, is target of FGF signaling and involved in induction of notochord, mesenchyme, and brain in ascidian embryos. Dev Biol 261:25–38
Nakatani Y, Nishida H (1994) Induction of notochord during ascidian embryogenesis. Dev Biol 166:289–299
Nakatani Y, Yasuo H, Satoh N, Nishida H (1996) Basic fibroblast growth factor induces notochord formation and the expression of As-T, a brachyury homolog, during ascidian embryogenesis. Development 122:2023–2031
Nishida H (1987) Cell lineage analysis in ascidian embryos by intracellular injection of a tracer enzyme. III. Up to the tissue-restricted stage. Dev Biol 121:526–541
Nishida H (2005) Specification of embryonic axis and mosaic development in ascidians. Dev Dyn 233:1177–1193
Nishikata T, Yamada L, Mochizuki Y, Satou Y, Shin-i T, Kohara Y, Satoh N (2001) Profiles of maternally expressed genes in fertilized eggs of Ciona intestinalis. Dev Biol 238:315–331
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory, New York
Satoh N (2003) The ascidian tadpole larva: comparative molecular development and genomics. Nat Rev Genet 4:285–295
Satou Y, Satoh N (1999) Developmental gene activities in ascidian embryos. Curr Opin Genet Dev 9:542–547
Satou Y, Kusakabe T, Araki I, Satoh N (1995) Timing of initiation of muscle-specific gene expression in the ascidian embryo precedes that of developmental fate restriction in lineage cells. Dev Growth Differ 37:319–327
Satou Y, Takatori N, Fujiwara S, Nishikata T, Saiga H, Kusakabe T, Shin-i T, KoharaY, Satoh N (2002) Ciona intestinalis cDNA projects: expressed sequence tag analyses and gene expression profiles during embryogenesis. Gene 287:83–96
Silverman ES, Baron RM, Palmer LJ, Le L, Hallock A, Subramaniam V, Riese RJ, McKenna MD, Gu X, Libermann TA, Tugores A, Haley KJ, Shore S, Drazen JM, Weiss ST (2002) Constitutive and cytokine-induced expression of the ETS transcription factor ESE-3 in the lung. Am J Respir Cell Mol Biol 27:697–704
Takahashi H, Hotta K, Erives A, Di Gregorio A, Zeller RW, Levine M, Satoh N (1999) Brachyury downstream notochord differentiation in the ascidian embryo. Genes Dev 13:1519–1523
Tugores A, Le J, Sorokina I, Snijders AJ, Duyao M, Reddy PS, Carlée L, Ronshaugen M, Mushegian A, Watanaskul T, Chu S, Buckler A, Emtage S, McCormick MK (2001) The epithelium-specific ETS protein EHF/ESE-3 is a context-dependent transcriptional repressor downstream of MAPK signaling cascades. J Biol Chem 276:20397–20406
Wada S, Saiga H (2002) HrzicN, a new Zic family gene of ascidians, plays essential roles in the neural tube and notochord development. Development 129:5597–5608
Yagi K, Satou Y, Mazet F, Shimeld SM, Degnan B, Rokhsar D, Levine M, Kohara Y, Satoh N (2003) A genomewide survey of developmentally relevant genes in Ciona intestinalis. III. Genes for Fox, ETS, nuclear receptors and NFκB. Dev Genes Evol 213:235–244
Yagi K, Satou Y, Satoh N (2004) A zinc finger transcription factor, ZicL, is a direct activator of Brachyury in the notochord specification of Ciona intestinalis. Development 131:1279–1288
Yagi K, Takatori N, Satou Y, Satoh N (2005) Ci-Tbx6b and Ci-Tbx6c are key mediators of the maternal effect gene Ci-macho1 in muscle cell differentiation in Ciona intestinalis embryos. Dev Biol 282:535–549
Yamada L, Kobayashi K, Degnan B, Satoh N, Satou Y (2003) A genomewide survey of developmentally relevant genes in Ciona intestinalis. IV. Genes for HMG transcriptional regulators, bZip and GATA/Gli/Zic/Snail. Dev Genes Evol 213:245–253
Yang S-H, Bumpass DC, Perkins ND, Sharrocks AD (2002) The ETS domain transcription factor Elk-1 contains a novel class of repression domain. Mol Cell Biol 22:5036–5046
Yasuo H, Satoh N (1993) Function of vertebrate T gene. Nature 364:582–583
Acknowledgements
We would like to thank Z. Imoto at Usa Marine Biological Institute of Kochi University for collecting animals, and the staff of Usa Marine Biological Institute for their hospitality. We are grateful to T. Nishikata at Konan University, N. Satoh and Y. Satou at Kyoto University, and H. Takahashi at the National Institute for Basic Biology for providing the animals. This work was supported by a Grant-in-Aid for Scientific Research on Priority Areas ‘Genome Science’ from MEXT Japan and Asahi Glass Foundation to S.F.A.S. was supported by the Japan Science Society.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by N. Satoh
Rights and permissions
About this article
Cite this article
Anno, C., Satou, A. & Fujiwara, S. Transcriptional regulation of ZicL in the Ciona intestinalis embryo. Dev Genes Evol 216, 597–605 (2006). https://doi.org/10.1007/s00427-006-0080-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00427-006-0080-9