
Abstract
Main conclusion
Red light (RL) accelerated starch accumulation in S. polyrhiza, but higher protein content under blue light (BL) was associated with the upregulation of most DEGs enriched for specific GO terms and KEGG pathways.
Abstract
Red light (RL) and blue light (BL) greatly influence the growth and physiological processes of duckweed. Physiological and molecular mechanisms underlying the response of duckweed to different light qualities remain unclear. This study employed physiological and transcriptomic analyses on duckweed, Spirodela polyrhiza “5510”, to elucidate its differential response mechanisms under RL, BL, and white light conditions. Changes in growth indicators, ultrastructure alterations, metabolite accumulations, and differentially expressed genes (DEGs) were measured. The results showed that BL promoted both biomass and protein accumulations, while RL promoted starch accumulation. A total of 633, 518, and 985 DEGs were found in white-vs-red, white-vs-blue, and red-vs-blue comparison groups, respectively. In Gene Ontology (GO) enrichment analysis, the DEGs in all three comparison groups were significantly enriched in two GO terms, carboxylic acid metabolic process and lyase activity. In Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis, the DEGs were greatly enriched in two pathways, histidine metabolism and isoquinoline alkaloid biosynthesis. Higher protein content under BL was associated with the upregulation of most DEGs enriched with the GO terms and KEGG pathways. Furthermore, the light qualities influenced the gene expression patterns of other metabolic pathways, like carotenoid biosynthesis, and the regulation of these genes may explain the level of photosynthetic pigment content. The results revealed the physiological changes and transcriptome-level responses of duckweed to three light qualities, thereby providing bases for further research studies on the ability of duckweed as a biomass energy source.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With the rapid growth of world energy demand, the increasing reliance on fossil fuels will bring a series of challenges that affect sustainable development, especially energy shortages and environmental pollution. Biofuels, such as bioethanol and biomass production, have the potential to replace traditional fossil fuels and could reduce our current reliance on fossil fuels. At present, most bioethanol production involves bioenergy crops or agricultural residues with high proportions of starch or sugar, such as sugarcane, corn, cassava, and sweet potato (Sanchez and Cardona 2008; Papong and Malakul 2010). It may also pose other environmental problems while competing for agricultural land use mainly for food or feed crop production (Crutzen et al. 2007; Searchinger et al. 2008). Thus, it is urgent to find alternative sources of raw materials for bioethanol production (Cui and Cheng 2015).
As a small flowering aquatic plant with global adaptability, duckweed has become a new potential alternative aquatic bioenergy crop in recent years (Ge et al. 2012; Zhao et al. 2015a). Compared to most bioenergy plants, duckweed has a longer yearly production period as well as a faster growth rate. Previous studies have shown that duckweed can grow up to 12.4 g m−2 day−1 dry weight and yield up to 55 tons hectare−1 year−1 dry weight in areas with suitable temperatures (Chaiprapat et al. 2005; Xu et al. 2011). In recent years, duckweed has been successfully used for bioethanol production, and more significantly, the high starch and low lignin properties of duckweed make it an ideal industrial feedstock (Cui et al. 2011; Chen et al. 2012). In addition, duckweed can effectively remove nutrients from eutrophic wastewater during its growth process, indicating its utility for wastewater treatment (Cheng and Stomp 2009; Li et al. 2016; Zhong et al. 2016). Hence, growing duckweed for biofuels can also provide significant environmental benefits.
One of the essential characteristics of a bioenergy feedstock is its high starch percentage. The induction of starch accumulation in duckweed is a prerequisite for it to become a high-quality bioenergy feedstock. However, the starch content of duckweed is significantly affected by its growing conditions, including light as one of its very important environmental factors. The growth and metabolism of plants are significantly regulated by both light intensity and light quality (Zhong et al. 2018). The regulation of light intensity on plants has been well studied in the past (Artetxe et al. 2002). In contrast, the research on the molecular basis of light quality regulation of plant growth and metabolism is relatively lacking. Since the first genome assembly of Spirodela polyrhiza was published (Wang et al. 2014), the genome assembly has been improved using Pacbio or Oxford long reads by different research groups, respectively (Hoang et al. 2018; An et al. 2019; Harkess et al. 2021). These excellent available duckweed genomes provide an opportunity to analyze gene localization, gene classification, and functional annotation on the molecular basis of light quality on the regulation of growth and metabolism in duckweed (Acosta et al. 2021). In the present study, three light conditions under 60 μmol photons m−2 s−1 intensity (red 660 nm, blue 460 nm, and white light 380–800 nm) were examined to figure out their effects on the growth and metabolism of the duckweed strain Spirodela polyrhiza “5510”. The transcriptome profiling and differentially expressed genes (DEGs) involved in the regulation of duckweed growth and metabolism under three light wavelengths were also examined to determine the possible molecular mechanisms by which duckweed respond to different light wavelengths. This work aimed to reveal the molecular response of duckweed to three light qualities at the transcriptome level and provide bases for future research studies on the ability of duckweed as a biomass energy source.
Materials and methods
Plant materials and experimental design
Samples of Spirodela polyrhiza “5510” (international identification of duckweeds) were isolated from Chaohu Lake (31°39′12.4″, 117°43′38.3″) in China (Zhao et al. 2012). The duckweed samples were gently rinsed with 1% of sodium hypochlorite solution (NaClO) for 1 min to remove surface contaminants. Individual fronds were incubated in modified Schenk-Hildebrandt medium and investigated under non-sterile conditions (SH medium; Schenk and Hildebrandt 1972). The duckweed samples were cultivated in a growth chamber at 25 ± 0.5 °C with a diurnal cycle of 16/8 h day/night.
To establish the influences of three light wavelengths on the duckweed cultures, duckweed were cultivated in a growth chamber (LB-175 N, Longyue, Shanghai, China) incubated at 25 ± 0.5 °C, and irradiated with varied lighting wavelengths, specifically: red light wavelength at 660 ± 5 nm (RL), blue light wavelength at 460 ± 5 nm (BL), and white light wavelength at 380–800 nm (WL) produced by light-emitting diode (LED). Beakers were irradiated under 60 μmol photons m−2 s−1 with a 16/8 h day/night cycle. Duckweed cultures with 1 g initial fresh weight were inoculated in a 250 mL beaker with modified SH medium. The beaker used had a surface area of 36 cm2. The duckweeds were exposed to the three light treatments for 12 days, harvested at the end of the light treatment on day 12, and then used for all data measurements. All experiments were performed in triplicates. Evaporated water was replenished daily with ultrapure water.
Measurement of growth rate and pigments
The light-treated fresh duckweed cultures were harvested at the end of the light treatment on day 12 after removing the surface medium, then lyophilized at − 50 °C, and then weighed. Duckweed growth rate was calculated using the following equation (Naumann et al. 2007):
The measured parameter x (DW) at the end (tn) and the start of the experiment (t0) for each repetition separately, where xtn refers the dry weight of the harvested duckweed (g) at time t and xt0 stands for the dry weight of the duckweed at the start of the experiment (Eq. 1).
A fresh sample of duckweed was prepared by taking 0.2 g, and then was ground and added to 80% of acetone until the sample turned white. The sample was filtered, and the filtrate was added with distilled water to a total volume of 100 mL. The absorbance of the extract was measured at 470 nm, 646 nm, and 663 nm using a spectrophotometer (TU-1810s, PERSEE, Beijing, China). The levels of chlorophyll a (Chl a), chlorophyll b (Chl b), and carotenoids (Car) were measured using the formula below (Lichtenthaler 1987):
The Chl a, Chl b, and Car in the formula (Eqs. 2–4) presented in milligrams per liter.
Determination of starch and amino acid content
The duckweed samples were harvested at the end of the light treatment on day 12 and lyophilized, and the starch content of duckweed calculated using the formula: total sugar content (starch content) = glucose content × 0.909) (Zhang et al. 2011).
Dried duckweed powder was used to measure the amino acid content. Hydrolyzed samples were characterized following the European Community (98/64/EG) guidelines. Sixteen different amino acid samples were separated by ion-exchange chromatography, and the reaction with ninhydrin was tested at 570 nm (proline 440 nm) using an automated amino acid analyzer (Model LC-3000, Eppendorf Biotronik, Hamburg, Germany).
Transmission electron microscopy images
Samples of duckweed were taken at the end of the light treatment on day 12, and their ultrastructure was observed using transmission electron microscopy (TEM). Samples were approximately 5 mm, fixed using 2.5% of glutaraldehyde, and placed in 0.2 M sucrose and 0.1 M cacodylate buffer (pH 7.2) overnight, as described by Zhong et al. (2016). Samples were postfixed with 1% of osmium tetroxide for 4 h, dehydrated in graded acetone solutions, and then resin-embedded based according to the method of Zhong (2016). Each treatment group had four replicates, each replicate examined had two samples, and all were imaged at 80 kV with a TEM 1011 (JEOL Ltd, Tokyo, Japan). The ultrastructural analysis was reliable based on comparisons of similarity of individual treatment and repeat treatment images.
RNA extraction, cDNA library construction, and Illumina deep sequencing
Duckweed samples were taken at the end of the light treatment on day 12; all samples in the three treatments were collected and frozen immediately in liquid nitrogen in preparation for RNA extraction. Each treatment had six replicates. However, three replicates were combined as one sample, so each light treatment ended with two replicates samples. This was done to ensure that each RNA sample preparation is 1 μg, the required volume of total RNA input material. Total RNA was extracted using the Trizol kit (Invitrogen, Carlsbad, CA, USA), and then, DNA contaminants were removed using RNase-Free DNase I (TaKaRa, Shiga, Japan). RNA integrity was confirmed using agarose gel electrophoresis by a 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). The Qubit 2.0 spectrophotometer (Life Technology, Waltham, MA, USA) was used to determine the RNA concentrations. The oligo (dT) magnetic beads and a fragmentation buffer were used to purify and fragment the mRNA, respectively. To synthesize the first-strand cDNA, reverse transcriptase and hexamer primer were used, and then, DNA polymerase I and RNase H were added to synthesize the second-strand cDNA. Then, agarose gel electrophoresis and PCR amplification were employed to purify and enrich the DNA fragments, respectively. Finally, cDNA libraries were created and sequenced with the Illumina HiSeq™ 2000 platform.
Bioinformatic analysis
After sequencing, the acquired reads were filtered by removing the reads containing the adapters and low-quality bases. The genome assembly of the duckweed, S. polyrhiza (Wang et al. 2014) was employed as the reference for RNA-seq analysis. All clean reads were mapped to the reference using Hisat2 (Kim et al. 2015). Gene expression levels were estimated by fragments per kilobase of transcript per million fragments mapped (FPKM). Annotation and categorization of assembled transcripts were performed through BLAST searches in various protein databases, including Cluster of Orthologous Groups of proteins (COG), GO, KEGG, euKaryotic Orthologous Groups (KOG), Swiss-Prot, and NR database.
Differentially expression genes analysis
The DESeq R package (1.10.1) was performed to analyze differential expressions between two light treatments at a time. WL was compared to RL (white-vs-red) and BL (white-vs-blue), and third comparison was of RL and BL (red-vs-blue). DESeq offers statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The method of Benjamini and Hochberg was used to control the false discovery rate and to adjust the obtained P values. Adjusted P values < 0.01 and log2 (fold change) absolute values > 1 were designated as differentially expressed genes.
The distribution of single genes was determined by GO and KEGG enrichment analysis based on their functional and biological pathways. The clusterProfiler R package was performed to analyze the DEGs implemented in GO enrichment. Enrichment analysis uses hypergeometric tests to identify GO entries that are markedly enriched compared to the whole genomic background. KEGG enrichment analysis was performed using the KOBAS software to test the statistical enrichment of DEGs in KEGG pathways. Additionally, analyses using the clusterProfiler R package were used to find KEGG pathway that is significantly enriched compared to the whole genome background.
Statistical analysis
All experiments were performed with at least three replicates. Statistical analyses with SPSS software (SPSS Inc., Chicago, USA) were used. The significance of the experimental results was analyzed by ANOVA, where P < 0.05 was considered statistically significant.
Results
Effects of light wavelengths on duckweed growth
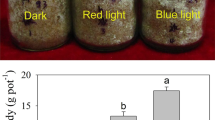
To evaluate the influences of light quality on growth rate, S. polyrhiza 5510 cultures were grown for 12 d under three light wavelength conditions. After 12 d, FW and DW of S. polyrhiza 5510 were measured. The growth rates (DW) were 0.146 ± 0.001 in RL, 0.147 ± 0.001 in BL, and 0.142 ± 0.001 in WL treatment conditions (Table 1). The growth rates of duckweeds under BL and RL were both higher than WL.
Table 1 also shows the contents of chlorophyll and carotenoid in S. polyrhiza 5510 grown under RL, BL, and WL conditions. In general, chlorophyll contents of duckweeds under the three light conditions showed no significant difference. However, BL-treated duckweed samples had higher carotenoid contents (129.82 ± 5.82 µg g−1) than the RL-treated and WL-treated samples measured at 88.87 ± 12.54 and 96.73 ± 4.13 (WL) μg g−1, respectively.
Effects of light wavelengths on starch and amino acid contents
Table 2 shows the varied influences of different light qualities on values of the starch and amino acid contents in S. polyrhiza. The results showed that the starch contents in S. polyrhiza were highest under RL treatment at 43.55 ± 2.88% (of DW). Such values were significantly higher by 50% compared to the WL treatment, while BL treatment showed increased starch contents by approximately 22% over the values under WL treatment. The total 16 amino acid contents in S. polyrhiza were highest under BL treatment (32.50 ± 0.04%), while values were 25.46 ± 0.04% in RL and 30.81 ± 0.03% in WL (Table 2).
The effect of different light qualities on the starch and amino acid contents can also be observed in the ultrastructure of S. polyrhiza fronds (Fig. 1). In RL treatment, chloroplasts contained many starch granules and darkly colored osmiophilic plastoglobuli in S. polyrhiza fronds (Fig. 1a). In contrast, chloroplasts exposed to BL included narrow and long-starch granules with small osmiophilic plastoglobuli (Fig. 1b), while small starch granules were observed in chloroplasts exposed to WL (Fig. 1c).

The ultrastructure of chloroplasts in S. polyrhiza 5510 under different light treatments. a Ultrastructure of chloroplasts in S. polyrhiza 5510 under red light treatment, b under blue light treatment, and c under white light treatment. Chl, chloroplast; S, starch grain; O, osmiophilic plastoglobuli
Sequencing data and DEGs analysis
Based on the growth and physiological characteristics of S. polyrhiza exposed to different wavelengths of light, transcriptome analysis was performed to show the influences of three light wavelengths on the expression of genes responsible for the observed physiological changes. All RNA-seq data are available at National Center for Biotechnology Information (NCBI) under project PRJNA760549. From six sequencing samples of three treatments (two biological duplicates for each treatment), an average of approximately 5.83 Gb clean reads for each sample was obtained. Each sample with a Q30 above 91.66% and GC content above 59.94% (Table 3), in the same treatment group showed that the correlations between the two biological replicates were all greater than 98% (Fig. 2a). Alignment of the clean reads to the S. polyrhiza reference genome showed matches at approximately 81.92 – 85.68% (Table S1). The mapped clean reads were used to estimate the gene expression level and detect the DEGs. The comparison of WL and RL treatments (white-vs-red) revealed 633 DEGs of which 263 genes were upregulated and 370 genes were downregulated (Fig. 2c). 518 DEGs were detected in the comparison of WL and BL treatments (white-vs-blue), and 219 and 299 genes were upregulated and downregulated, respectively (Fig. 2c). The comparison of RL and BL treatments (red-vs-blue) showed the highest DEGs at 985 genes of which 471 upregulated and 514 downregulated genes were observed (Fig. 2c). A total of 37 DEGs were detected in all three treatment comparisons, while the most special DEGs (444) were found in the RL-BL comparison group (red-vs-blue, Fig. 2b).

a Correlation between samples of S. polyrhiza 5510 under different light treatments. b Venn diagram of white-vs-red, white-vs-blue, and red-vs-blue. c Volcano plot of DEGs of white-vs-red, white-vs-blue, and red-vs-blue
GO and KEGG pathway enrichment analysis of DEGs
To classify the manifestations of DEGs, the GO enrichment analysis was performed to determine what biological processes (BP), cellular components (CC), and molecular functions (MF) were the DEGs are overrepresented in. Based on the GO database, GO analysis of white-vs-red, white-vs-blue, and red-vs-blue revealed a total of 360, 266, and 573 DEGs, respectively (Table S2) and were enriched in 7, 2, and 9 GO terms, respectively (Table 4). In the BP category, DEGs were grouped into metabolic process, cellular process, and biological regulation. In the CC category, DEGs were mainly categorized as membrane, cell, and cell part. In the MF category, DEGs were enriched in binding and catalytic activity (Fig. 3).

GO enrichment analysis of DEGs of S. polyrhiza 5510 under different light treatments. The left Y-axis shows the GO term. The X-axis shows the proportion of genes of interest in the entry to the number of all DEGs. The color of the dot represents P value; the size of the dot represents the number of DEGs annotated in the GO term
The KEGG pathway analysis revealed 407 (white-vs-red), 308 (white-vs-blue), and 663 (red-vs-blue) DEGs in the comparison groups (Table S2). Figure 4 and Table 5 show the results of KEGG pathway analysis performed in the three comparison groups of the three light treatments. In white-vs-red, 225 genes were enriched in 85 pathways, of which 82 genes were significantly enriched in nine pathways (Table 5). In white-vs-blue, 193 genes were enriched in 78 pathways, of which 111 genes were significantly enriched in 11 pathways (Table 5). In red-vs-blue, 368 genes were enriched in 96 pathways, of which 152 genes were significantly enriched in 16 pathways (Table 5).

KEGG pathway enrichment analysis of S. polyrhiza 5510 under different light treatments. The left Y-axis shows the KEGG pathway. The X-axis shows the Rich factor. The color of the dot represents Q value, the size of the dot represents the number of DEGs annotated in the pathway, and the shape of the dot represents the type of DEGs in the pathway
Discussion
Photosynthesis is a unique biological pathway through which plants convert active light energy into stable chemical energy. Light quality has a deep impact on photosynthetic productivity and various physiological activities of plants (Neff et al. 2000). Plants can effectively absorb blue and red light in the spectrum, resulting in higher photosynthetic efficiency compared to other spectra. The use of an appropriate proportion of blue and red light at the same time has been reported as the best lighting strategy for most plants (Xu et al. 2019). For example, the total biomass yield of S. polyrhiza increased significantly under monochromatic (blue: 450 nm; red: 660 nm) and tailored light conditions (simultaneous red and blue light) compared with duckweeds grown under a fluorescent lamp (Xu et al. 2019). This study revealed similar higher growth rate observed in duckweed cultures under RL and BL treatments (Table 1). The same result was observed in Lactuca sativa L. at low light intensities (Muneer et al. 2014). Short wavelengths like BL with the highest maximum specific growth rates (μ max) can produce higher photosynthetic efficiency, while long wavelengths like RL lead to lower photosynthesis efficiency. Thus, from a per light quantity perspective, BL has a higher photosynthetic efficiency than RL or WL, resulting in a higher growth rate (Das et al. 2011). In addition, anti-reactive oxygen species values are higher in plants exposed to BL that helps plants to resist oxidative stress, which may be another reason for high biomass accumulation (Hideg et al. 2013).
Chlorophyll and carotenoid play important roles in plant light harvesting and photoprotection. Complementary chromatic adaptation modulates the relative content of the major light-harvesting pigments, so that the absorption spectrum of the major light-harvesting pigments is similar to that of the incident light. Complementary chromatic adaptation may be one of the reasons for the higher content of chlorophylls and carotenoids in BL (Mouget et al. 2005). The maximum absorption of chlorophyll a and b in the red region of the visible spectrum is 663 and 642 nm, respectively, while the maximum absorption in the blue region is 430 and 453 nm, respectively. Furthermore, lutein and β-carotene among carotenoids had the highest absorption at 448 and 454 nm (Lefsrud et al. 2008). Our findings were consistent with a previous report about total chlorophyll and carotenoid levels accumulated under BL (Kopsell and Sams 2013). Moreover, a study on barley leaves under RL irradiation has shown abnormal chloroplasts’ structure and decreased chlorophyll contents, but the abnormality was reversed under BL irradiation and chlorophyll contents were relatively stable (Voskresenskaya 1972). In our study, the chloroplasts’ ultrastructure of S. polyrhiza was strongly influenced by the three light qualities (Fig. 1). Under RL treatment, the chloroplasts showed various shapes, different sizes, and severe malformation. Additionally, the boundary between the grana thylakoid and stromal thylakoid was not clear, with gaps and disorganization among grana lamellae (Fig. 1a). Under BL treatment, chloroplasts were crescent-shaped with well-developed thylakoids, of which many thylakoids were closely packed in the neatly arranged granum of chloroplasts. Furthermore, the grana thylakoid and stromal thylakoid were well connected with clear borders (Fig. 1b). Under WL treatment, the chloroplasts were elliptical in shape. The grana thylakoid and stromal thylakoid were clearly visible with gaps inside the granum (Fig. 1c). Moreover, we observed the largest number of chloroplasts per cell and the thickest granules in BL-treated duckweed cultures. Therefore, chloroplasts developed well under BL treatment, and in general, light quality significantly affected the development of chloroplasts.
Recent studies have shown that light wavelengths not only affect plant growth and development but also influence the levels of metabolites. In this study, the content of starch in S. polyrhiza was significantly highest under RL treatment (Table 2) that is consistent with the results of studies in peanut and potato plants (Chen et al. 2018a; Li et al. 2018). The accumulation of starch and soluble sugars in leaves under RL is caused by its inhibition of the output of photosynthetic products. Although duckweed cultures under RL treatment accumulated more starch, the highest biomass yield was observed in cultures under BL treatment. This phenomenon may be due to the plant’s inability to transport its photosynthetic products efficiently.
Chloroplasts are important organelles for energy conversion as well as sites of many important metabolic reactions, such as biosynthesis of amino acids, starch, and fatty acids (van Wijk 2000; Baginsky and Gruissem 2004). In this study, the ultrastructure of S. polyrhiza under RL treatment showed the most volume of chloroplasts occupied by starch granules that influenced the grana lamellae’s function. The grana lamellae were decentralized and unstacked, and the net photosynthetic rate was lowest. Starch accumulation in chloroplasts affects their normal function, reducing the photosynthetic rate, in particular Bondada and Syvertsen (2005) and Li et al. (2018). Under BL treatment, the S. polyrhiza cultures have small starch grains and uniform grana lamellae, of which both phenomena are relative to the high net photosynthetic rate obtained under BL.
RL induces transformation of the phytochrome system, while BL affects the chlorophyll formation, photomorphogenesis, and stomatal opening (Chen et al. 2018b). Light quality significantly affects the biosynthesis and accumulation of carbohydrate and protein. In pea seedlings, RL significantly enhanced the biosynthesis of soluble sugars, but inhibited the biosynthesis of soluble proteins, whereas BL served to promote protein biosynthesis and to protect against protein degradation (Chen et al. 2018b). The content of amino acids would fluctuate widely with the light quality. In maize grown under BL, the content of amino acids was enhanced and was much higher than values observed under RL or dark treatments. Moreover, narrow bandwidth blue light was shown to induce amino acid production in rice seedling leaves under different LED light sources (Jung et al. 2013).
The important roles of functional genes or metabolic pathways in transcriptional regulation can be elucidated by the annotations of different genes in different transcriptomes. To investigate the potential role of DEGs in the regulation of metabolic pathways by spectroscopy light wavelength, the GO classification and KEGG pathways were analyzed through three comparison groups.
In the GO enrichment analysis, DEGs in the white-vs-red group were significantly enriched on seven GO terms (Table 4). Biological processes (BP) were mainly enriched in three pathways: carboxylic acid metabolic process, response to auxin, and regulation of transcription, DNA-templated. Cellular components (CC) were mainly concentrated on anaphase-promoting complex. Molecular functions (MF) were mainly focused on three pathways: calcium ion binding, carbohydrate binding, and lyase activity. The DEGs in white-vs-blue were significantly enriched on two GO terms (Table 4). BP were mainly enriched in carboxylic acid metabolic process, while MF were mainly focused on lyase activity. The DEGs in red-vs-blue were significantly enriched on nine GO terms (Table 4). BP were mainly enriched in two pathways: negative regulation of growth, and carboxylic acid metabolic process. CC were mainly concentrated on anaphase-promoting complex. MF were mainly focused on six pathways: transferase activity including transferring acyl groups other than amino-acyl groups, carbohydrate binding, lyase activity, copper ion binding, coenzyme binding, and hydrolase activity acting on ester bonds.
Moreover, we focused on the carboxylic acid metabolic process (GO:0,019,752) term in the BP category and found four DEGs present in the three comparison groups. Additionally, the regulation of these four DEGs in white-vs-red was opposite in both white-vs-blue and red-vs-blue comparisons. These four DEGs were grouped in the serine decarboxylase-like (Table S3). Serine decarboxylase-like and amino acid aldolases convert phenylalanine, methionine, leucine, and tryptophan into PHA, 4-(methylthio) butanal, 3-methylbutanal, and indole-3-acetaldehyde (Torrens-Spence et al. 2014). The Spipo4G0109700.v2 and Spipo4G0110000.v2 were upregulated under RL treatment (white-vs-red and red-vs-blue); therefore, the contents of phenylalanine and leucine were reduced, while the methionine content increased (Table 2).
In the molecular functions (MF) category, we focused on lyase activity (GO:0,016,829) term and found three (white-vs-red) downregulated DEGs, three (white-vs-blue) upregulated DEGs, and five (red-vs-blue) upregulated DEGs. Moreover, all DEGs were clustered within the terpene synthase (Table S3). The expression level of terpene synthase has been shown to react under BL and RL treatments (Fu et al. 2015). In plants, terpenoids play crucial roles in physiological and biochemical processes, such as photosynthesis and developmental regulation (Pichersky and Raguso 2018). In the present study, higher expression levels of terpene synthases under BL treatment may be responsible for better photosynthesis and development.
Based on the KEGG analysis in the three comparison groups, the transcripts were greatly enriched in two protein synthesis-associated pathways, namely, histidine metabolism and isoquinoline alkaloid biosynthesis (Table 5). Notably, the pathway showed opposite regulatory trends in BL and RL treatments. Within the histidine metabolism pathway, three DEGs were present in all three comparison groups. In white-vs-red, two of three DEGs (Spipo4G0109700.v2 and Spipo4G0110000.v2) were upregulated and annotated as serine decarboxylase-like. The one downregulated DEG (Spipo0G0176400.v2) was annotated as pyridoxal phosphate (PLP)-dependent transferases superfamily protein (Table S4). Inversely, Spipo4G0109700.v2 and Spipo4G0110000.v2 were downregulated in white-vs-blue, while Spipo0G0176400.v2 was upregulated. An Arabidopsis study has shown that serine decarboxylase catalyzed the conversion of serine to ethanolamine and that overexpression of serine decarboxylase reduced the level of amino acid compounds arising from the mitochondrial tricarboxylic acid cycle (Liu et al. 2018). In our study, the serine and amino acid contents at low and high levels in the RL and BL treatments, respectively, may be due to the serine decarboxylase-like observed high and low expression levels under RL and BL treatments, respectively (Table 2). Meanwhile, PLP-dependent transferase superfamily, including all group II decarboxylases of histidine, l-tyrosine, glutamate and a variety of aromatic amino acids, and sphingosine phosphate cleavage enzymes (Kezmarsky et al. 2005), is another possible reason for the amino acid content variations in our study. Within the isoquinoline alkaloid biosynthesis pathway, two DEGs were present in all three comparison groups. Two downregulated DEGs (Spipo25G0004100.v2 and Spipo1G0042600.v2) in white-vs-red were annotated as polyphenol oxidase and tyrosine/DOPA decarboxylase 2-like, respectively, but these DEGs were upregulated in the white-vs-blue group (Table S4). The phenolic amino acid tyrosine could be oxidized by polyphenol oxidase, and tyrosine/DOPA decarboxylase catalyzes the conversion of L-tyrosine to dopamine in early steps of benzylisoquinoline alkaloid biosynthesis pathway (Yoruk and Marshall 2003; Madani et al. 2019). Therefore, the upregulation of Spipo25G0004100.v2 and Spipo1G0042600.v2 under BL treatment (white-vs-blue and red-vs-blue) might be due to reduced tyrosine content (Table 2).
Based on the results of the KEGG analysis, it showed that only BL treatment affected some pathways. For example, carotenoid biosynthesis pathway was significantly aggregated in white-vs-blue and red-vs-blue, but no aggregation in white-vs-red. Through carotenoid biosynthesis pathway analysis, the expression of two key enzymes were found upregulated in the synthesis of carotenoid, namely, the expression levels of transcripts encoding violaxanthin de-epoxidase (EC:1.23.5.1) and 9-cis-epoxycarotenoid dioxygenase (NCED) (EC:1.13.11.51) (Fig. 5).

Expression patterns of carotenoid biosynthesis-related transcripts of S. polyrhiza 5510. Red boxes indicate upregulated enzymes; blue means mix regulated enzymes
Violaxanthin de-epoxidase is a key enzyme in the luteinizing cycle of the carotenoid biosynthetic pathway that catalyzes the conversion of violaxanthin to zeaxanthin through antheraxanthin. Zeaxanthin and antheraxanthin can divert chlorophyll excess energy that is dissipated as heat and by scavenging ROS, thus protecting the photosynthetic apparatus from photodamage (Hager and Perz 1970; Xu et al. 2016).
NCED is the rate-limiting enzyme in the abscisic acid (ABA) biosynthesis. The carbon skeleton of abscisic acid is similar to the terminal part of some carotenoids, so the pathway of ABA production from oxidative breakdown of carotenoids is an indirect pathway for ABA synthesis. Related research has indicated that the early stages of the carotenoid biosynthesis pathway during the ripening of ‘Navelate’ orange pericarp are severely blocked, thereby reducing ABA levels (Romero et al. 2019). In this study, the increased expressions of violaxanthin de-epoxidase and NCED under BL treatment may have improved the resilience of duckweed and protected its photosynthetic system that resulted in higher biomass.
Conclusion
This study investigated the physiological effects of three types of light wavelengths on the growth and development of duckweed, S. polyrhiza 5510. The RL treatment accelerated starch accumulation in S. polyrhiza compared with BL and WL treatments. Higher protein content observed under BL was associated with the upregulation of most DEGs enriched for specific GO terms and KEGG pathways. In addition, the three light qualities induced different gene expression patterns of other metabolic pathways, such as carotenoid biosynthesis, and the regulation of these genes may explain the level of photosynthetic pigment content. Our results reveal the response of duckweed to different light qualities at the transcriptome level and provide bases for further studies on the ability of duckweed as a biomass energy source.
Author contributions statement
YZ and XLD conceived and designed the experiments. YZ, LW, and ZMM performed the experiments and analyzed the data. YZ, LW, ZMM, and XLD wrote the paper.
Data availability
All RNA-seq data are available at National Center for Biotechnology Information (NCBI) under project PRJNA760549.
Abbreviations
- BL:
-
Blue light
- DEG:
-
Differentially expressed gene
- RL:
-
Red light
- WL:
-
White light
References
Acosta K, Appenroth KJ, Borisjuk L et al (2021) Return of the Lemnaceae: duckweed as a model plant system in the genomics and postgenomics era. Plant Cell 33:3207–3234. https://doi.org/10.1093/plcell/koab189
An D, Zhou Y, Li C, Xiao Q, Wang T, Zhang Y, Wu Y, Li Y, Chao DY, Messing J, Wang W (2019) Plant evolution and environmental adaptation unveiled by long-read whole-genome sequencing of Spirodela. Proc Natl Acad Sci USA 116(38):18893–18899. https://doi.org/10.1073/pnas.1910401116
Artetxe U, García-Plazaola JI, Hernández A, Becerril JM (2002) Low light grown duckweed plants are more protected against the toxicity induced by Zn and Cd. Plant Physiol Biochem 40(10):859–863. https://doi.org/10.1016/S0981-9428(02)01446-8
Baginsky S, Gruissem W (2004) Chloroplast proteomics: potentials and challenges. J Exp Bot 55(400):1213–1220. https://doi.org/10.1093/jxb/erh104
Bondada BR, Syvertsen JP (2005) Concurrent changes in net CO2 assimilation and chloroplast ultrastructure in nitrogen deficient citrus leaves. Environ Exp Bot 54(1):41–48. https://doi.org/10.1016/j.envexpbot.2004.05.005
Chaiprapat S, Cheng JJ, Classen JJ, Liehr SK (2005) Role of internal nutrient storage in duckweed growth for swine wastewater treatment. Transactions ASAE. 48(6):2247–2258. https://doi.org/10.13031/2013.20088
Chen Q, Jin Y, Zhang G, Fang Y, Xiao Y, Zhao H (2012) Improving production of bioethanol from duckweed (Landoltia punctata) by pectinase pretreatment. Energies 5(8):3019–3032. https://doi.org/10.3390/en5083019
Chen Y, Zhou B, Li J, Tang H, Tang J, Yang Z (2018b) Formation and change of chloroplast-located plant metabolites in response to light conditions. Int J Mol Sci 19(3):654. https://doi.org/10.3390/ijms19030654
Chen L, Xue X, Yang Y, Chen F, Zhao J, Wang X, Khan A, Hu Y (2018a) Effects of red and blue LEDs on in vitro growth and microtuberization of potato single-node cuttings. Front Agric Sci Eng. 5(2):197–205. https://doi.org/10.15302/J-FASE-2018224
Cheng JJ, Stomp AM (2009) Growing duckweed to recover nutrients from wastewaters and for production of fuel ethanol and animal feed. Clean: Soil, Air, Water. 37(1):17–26. https://doi.org/10.1002/clen.200800210
Crutzen PJ, MosierAR, SmithK A, Winiwarter W (2007) N2O release from agro-biofuel production negates global warming reduction by replacing fossil fuels. Atmos Chem Phys Discussions 7(4):11191–11205. https://hal.archives-ouvertes.fr/hal-00303019
Cui W, Cheng JJ (2015) Growing duckweed for biofuel production: a review. Plant Biol 17:16–23. https://doi.org/10.1111/plb.12216
Cui W, Xu J, Cheng JJ, Stomp AM (2011) Starch accumulation in duckweed for bioethanol production. Biol Engin Transactions 3(4):187–197. https://doi.org/10.13031/2013.37123
Das P, Lei W, Aziz SS, Obbard JP (2011) Enhanced algae growth in both phototrophic and mixotrophic culture under blue light. Bioresource Technol 102(4):3883–3887. https://doi.org/10.1016/j.biortech.2010.11.102
Fu X, Chen Y, Mei X, Katsuno T, Kobayashi E, Dong F, Watanabe N, Yang Z (2015) Regulation of formation of volatile compounds of tea (Camellia sinensis) leaves by single light wavelength. Sci Rep 5(1):16858. https://doi.org/10.1038/srep16858
Ge X, Zhang N, Phillips GC, Xu J (2012) Growing Lemna minor in agricultural wastewater and converting the duckweed biomass to ethanol. Bioresource Technol 124:485–488. https://doi.org/10.1016/j.biortech.2012.08.050
Hager A, Perz H (1970) Changes in the light-absorption of a carotenoid in an enzyme (de-epoxidase)-substrate (Violaxanthin)-complex. Planta 93:314–322. https://doi.org/10.1007/BF00384105
Harkess A, McLaughlin F, Bilkey N, Elliot K, Emenecker R, Mattoon E, Miller K, Czymmek K, Vierstra R, Meyers BC et al (2021) Improved Spirodela polyrhiza genome and proteomic analyses reveal a conserved chromosomal structure with high abundance of chloroplastic proteins favoring energy production. J Exp Bot 72:2491–2500. https://doi.org/10.1093/jxb/erab006
Hideg É, Jansen MA, Strid Å (2013) UV-B exposure, ROS, and stress: inseparable companions or loosely linked associates? Trends Plant Sci 18(2):107–115. https://doi.org/10.1016/j.tplants.2012.09.003
Hoang PTN, Michael TP, Gilbert S, Chu P, Motley ST, Appenroth KJ, Schubert I, Lam E (2018) Generating a high-confidence reference genome map of the Greater Duckweed by integration of cytogenomic, optical mapping, and Oxford Nanopore technologies. Plant J 96:670–684. https://doi.org/10.1111/tpj.14049
Jung ES, Lee S, Lim SH, Ha SH, Liu KH, Lee CH (2013) Metabolite profiling of the short-term responses of rice leaves (Oryza sativa cv. Ilmi) cultivated under different LED lights and its correlations with antioxidant activities. Plant Sci 210:61–69. https://doi.org/10.1016/j.plantsci.2013.05.004
Kezmarsky ND, Xu H, Graham DE, White RH (2005) Identification and characterization of a L-tyrosine decarboxylase in Methanocaldococcus jannaschii. Biochim Biophys Acta 1722(2):175–182. https://doi.org/10.1016/j.bbagen.2004.12.003
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4):357–360. https://doi.org/10.1038/NMETH.3317
Kopsell DA, Sams CE (2013) Increases in shoot tissue pigments, glucosinolates, and mineral elements in sprouting broccoli after exposure to short-duration blue light from light emitting diodes. J Amer Soc Hort Sci 138(1):31–37. https://doi.org/10.21273/JASHS.138.1.31
Lefsrud MG, Kopsell DA, Sams CE (2008) Irradiance from distinct wavelength light-emitting diodes affect secondary metabolites in kale. HortScience 43(7):2243–2244. https://doi.org/10.21273/HORTSCI.43.7.2243
Li Y, Zhang F, Daroch M, Tang J (2016) Positive effects of duckweed polycultures on starch and protein accumulation. Biosci Rep 36(5):e00380. https://doi.org/10.1042/BSR20160158
Li C, Liu D, Li L, Hu S, Xu Z, Tang C (2018) Effects of light-emitting diodes on the growth of peanut plants. Agron J 110(6):2369–2377. https://doi.org/10.2134/agronj2017.11.0674
Lichtenthaler HK (1987) Chlorophylls and carotenoids: pigments of photosynthetic biomembrane. Methods Enzymol 148:350–382. https://doi.org/10.1016/0076-6879(87)48036-1
Liu YC, Gunawan F, Yunus IS, Nakamura Y (2018) Arabidopsis serine decarboxylase 1 (SDC1) in phospholipid and amino acid metabolism. Frontiers Plant Sci 9:972. https://doi.org/10.3389/fpls.2018.00972
Madani H, Hosseini B, Karimzadeh G, Rahimi A (2019) Enhanced thebaine and noscapine production and modulated gene expression of tyrosine/dopa decarboxylase and salutaridinol 7-O-acetyltransferase genes in induced autotetraploid seedlings of Papaver bracteatum Lindl. Acta Physiol Plant 41(12):1–12. https://doi.org/10.1007/s11738-019-2984-9
Mouget JL, Rosa P, Vachoux C, Tremblin G (2005) Enhancement of marennine production by blue light in the diatom Haslea ostrearia. J Appl Phycol 17(5):437–445. https://doi.org/10.1007/s10811-005-0561-7
Muneer S, Kim EJ, Park JS, Lee JH (2014) Influence of green, red and blue light emitting diodes on multiprotein complex proteins and photosynthetic activity under different light intensities in lettuce leaves (Lactuca sativa L.). Int J Mol Sci 15(3):4657–4670. https://doi.org/10.3390/ijms15034657
Naumann B, Eberius M, Appenroth KJ (2007) Growth rate based dose-response relationships and EC-values of ten heavy metals using the duckweed growth inhibition test (ISO 20079) with Lemna minor L. clone St. J Plant Physiol 164:1656–1664. https://doi.org/10.1016/j.jplph.2006.10.011
Neff MM, Fankhauser C, Chory J (2000) Light: an indicator of time and place. Genes Dev 14(3):257–271. https://doi.org/10.1101/gad.14.3.257
Papong S, Malakul P (2010) Life-cycle energy and environmental analysis of bioethanol production from cassava in Thailand. Bioresource Technol 101(1):S112–S118. https://doi.org/10.1016/j.biortech.2009.09.006
Pichersky E, Raguso RA (2018) Why do plants produce so many terpenoid compounds? New Phytol 220(3):692–702. https://doi.org/10.1111/nph.14178
Romero P, Lafuente MT, Rodrigo MJ (2019) A sweet orange mutant impaired in carotenoid biosynthesis and reduced ABA levels results in altered molecular responses along peel ripening. Sci Rep 9(1):9813. https://doi.org/10.1038/s41598-019-46365-8
Sanchez OJ, Cardona CA (2008) Trends in biotechnological production of fuel ethanol from different feedstocks. Bioresource Technol 99(13):5270–5295. https://doi.org/10.1016/j.biortech.2007.11.013
Schenk RU, Hildebrandt AC (1972) Medium and techniques for induction and growth of monocotyledonous and dicotyledonous plant cell cultures. Can J Bot 50(1):199–204. https://doi.org/10.1139/b72-026
Searchinger T, Heimlich R, Houghton RA, Dong F, Elobeid A, Fabiosa J, Tokgoz S, Hayes D, Yu TH (2008) Use of US croplands for biofuels increases greenhouse gases through emissions from land-use change. Science 319(5867):1238–1240. https://doi.org/10.1126/science.1151861
Torrens-Spence MP, von Guggenberg R, Lazear M, Ding H, Li J (2014) Diverse functional evolution of serine decarboxylases: identification of two novel acetaldehyde synthases that uses hydrophobic amino acids as substrates. BMC Plant Biol 14(1):247. http://www.biomedcentral.com/1471-2229/14/247
van Wijk KJ (2000) Proteomics of the chloroplast: experimentation and prediction. Trends Plant Sci 5(10):420–425. https://doi.org/10.1016/S1360-1385(00)01737-4
Voskresenskaya NP (1972) Blue light and carbon metabolism. Annu Rev Plant Physiol 123(1):219–234
Wang W, Haberer G, Gundlach H et al (2014) The Spirodela polyrhiza genome reveals insights into its neotenous reduction fast growth and aquatic lifestyle. Nat Commun 5:3311. https://doi.org/10.1038/ncomms4311
Xu J, Cui W, Cheng JJ, Stomp AM (2011) Production of high-starch duckweed and its conversion to bioethanol. Biosystems Engin 110(2):67–72. https://doi.org/10.1016/j.biosystemseng.2011.06.007
Xu J, Li Z, Yang H, Yang X, Chen C, Li H (2016) Genetic diversity and molecular evolution of a violaxanthin de-epoxidase gene in maize. Frontiers Genetics 7:00131. https://doi.org/10.3389/fgene.2016.00131
Xu YL, Tan L, Guo L, Yang GL, Li Q, Lai F, He KZ, Jin YL, Du A, Fang Y, Zhao H (2019) Increasing starch productivity of Spirodela polyrhiza by precisely control the spectral composition and nutrients status. Ind Crops Prod 134:284–291. https://doi.org/10.1016/j.indcrop.2019.04.002
Yoruk R, Marshall MR (2003) Physicochemical properties and function of plant polyphenol oxidase: a review. J Food Biochem 27(5):361–422. https://doi.org/10.1111/j.1745-4514.2003.tb00289.x
Zhang L, Zhao H, Gan M, Jin Y, Gao X, Chen Q, Guan J, Wang Z (2011) Application of simultaneous saccharification and fermentation (SSF) from viscosity reducing of raw sweet potato for bioethanol production at laboratory, pilot and industrial scales. Bioresource Technol 102(6):4573–4579. https://doi.org/10.1016/j.biortech.2010.12.115
Zhao Z, Shi HJ, Wang ML, Cui L, Zhao H, Zhao Y (2015) Effect of nitrogen and phosphorus deficiency on transcriptional regulation of genes encoding key enzymes of starch metabolism in duckweed (Landoltia punctata). Plant Physiol Biochem 86:72–81. https://doi.org/10.1016/j.plaphy.2014.11.007
Zhao H, Appenroth KJ, Landesman L, A. Salmea´n A, Lam E (2012) Duckweed rising at Chengdu: summary of the 1st International Conference on Duckweed Application and Research. Plant Mol Biol 78:627–632. https://doi.org/10.1007/s11103-012-9889-y
Zhong Y, Li Y, Cheng JJ (2016) Effects of selenite on chlorophyll fluorescence, starch content and fatty acid in the duckweed Landoltia punctata. J Plant Res 129(5):997–1004. https://doi.org/10.1007/s10265-016-0848-6
Zhong Y, Jin P, Cheng JJ (2018) A comprehensive comparable study of the physiological properties of four microalgal species under different light wavelength conditions. Planta 248(2):489–498. https://doi.org/10.1007/s00425-018-2899-5
Acknowledgements
This research was mainly funded by the China Postdoctoral Science Foundation (2019TQ0116).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Communicated by Dorothea Bartels.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhong, Y., Wang, L., Ma, Z. et al. Physiological responses and transcriptome analysis of Spirodela polyrhiza under red, blue, and white light. Planta 255, 11 (2022). https://doi.org/10.1007/s00425-021-03764-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00425-021-03764-4