
Abstract
Main conclusion
Plastid-based MNEI protein mutants retain the structure, stability and sweetness of their bacterial counterparts, confirming the attractiveness of the plastid transformation technology for high-yield production of recombinant proteins.
The prevalence of obesity and diabetes has dramatically increased the industrial demand for the development and use of alternatives to sugar and traditional sweeteners. Sweet proteins, such as MNEI, a single chain derivative of monellin, are the most promising candidates for industrial applications. In this work, we describe the use of tobacco chloroplasts as a stable plant expression platform to produce three MNEI protein mutants with improved taste profile and stability. All plant-based proteins were correctly expressed in tobacco chloroplasts, purified and subjected to in-depth chemical and sensory analyses. Recombinant MNEI mutants showed a protein yield ranging from 5% to more than 50% of total soluble proteins, which, to date, represents the highest accumulation level of MNEI mutants in plants. Comparative analyses demonstrated the high similarity, in terms of structure, stability and function, of the proteins produced in plant chloroplasts and bacteria. The high yield and the extreme sweetness perceived for the plant-derived proteins prove that plastid transformation technology is a safe, stable and cost-effective production platform for low-calorie sweeteners, with an estimated production of up to 25–30 mg of pure protein/plant.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
MNEI is a single chain derivative of monellin, one of the seven members of the “sweet proteins” family (Picone and Temussi 2012). These are structurally and biosynthetically unrelated plant proteins, provided with the remarkable feature of eliciting a sweet taste response in humans (Temussi 2011a). Recently, sweet proteins have received renewed attention from food, beverages and pharmaceutical companies looking for safe alternatives to sugar and traditional sweeteners (Kant 2005). The first sweet proteins to be characterized were thaumatin, from Thaumatococcus daniellii (van der Wel and Loeve 1972), and monellin, from Dioscoreophyllum cumminsii (Inglett and May 1969; Morris and Cagan 1972). Both proteins are incredibly sweet, in the order of 100,000 sweeter than sucrose on a molar basis (Inglett and May 1969). Sweet proteins taste is linked to their three-dimensional shape, which allows them to interact with the T1R2-T1R3 sweet taste receptor through a complex patch of complementary charged residues. To date, the model providing the best rationalization of such interaction is the “wedge model”, according to which sweet proteins bind to a cleft formed by portions of both subunits of the sweet taste receptors (Morini et al. 2005; Tancredi et al. 2004; Temussi 2011b, 2002). Sweet proteins need to be correctly folded to elicit the sweet taste response, which is why natural monellin loses its sweetness when heated above 50 °C. The protein indeed occurs as a small (~ 12 kDa), globular, heterodimer whose subunits, held together by non-covalent interactions and assembled in a cistatin-like fold (Murzin 1993; Ogata et al. 1987) dissociate at this temperature, so that the protein fold is lost. This physical limit has restricted the use of natural monellin as a sweetener, since industrial settings for food processing make often use of harsher conditions. In the attempt to overcome these limitations, single chain derivatives of monellin have been engineered, namely SCM and MNEI, through direct linkage of the two polypeptide chains or by introduction of the Gly-Phe dipeptide, respectively (Kim et al. 1989; Tancredi et al. 1992), that can only be produced through biotechnology. These “first generation” mutants possess the structure and sweetness of the parent protein and, additionally, have higher thermal stability and can correctly refold, regaining sweetness, after thermal denaturation at acidic pH. MNEI seems, therefore, particularly promising for industrial applications for several reasons: the well-characterized structure (Spadaccini et al. 2001; Hobbs et al. 2007; Spadaccini et al. 2016); a taste profile that, unlike many synthetic sweeteners, closely resembles that of sucrose (Di Monaco et al. 2013, 2014); and improved biochemical properties (e.g., increased thermal stability and retained function even after short boiling at acidic pH) compared to native monellin (Kim et al. 1989; Spadaccini et al. 2001). Moreover, recent studies have been undertaken to characterize the behavior of MNEI in more complex, application-oriented, matrices, such as agar-based or dairy gels (Miele et al. 2017a, b), and to begin the description of the physiological and eco-toxicological effects of its introduction in the food chain (Rega et al. 2017). The intrinsic structural limits of wild type monellin and the restricted availability of natural sources have led to pursue biotechnological approaches to produce sweet proteins, exploiting a variety of bacterial, yeast and plant hosts (Kim and Lim 1996; Lamphear et al. 2005; Masuda and Kitabatake 2006; Chen et al. 2007; Sun et al. 2007; Chen et al. 2011; Pham et al. 2012; Leone et al. 2015; Reddy et al. 2015; Cai et al. 2016; Kaul et al. 2018). Genetic engineering has in fact allowed for functional improvements of the original protein and its single chain derivatives, through the introduction of point mutations linked to taste (such as, for MNEI, Y65R or Q28K) or stability (E23Q, E23A, C41S) enhancements, or both (Esposito et al. 2006; Rega et al. 2015; Leone et al. 2016; Leone and Picone 2016; Liu et al. 2016; Pica et al. 2018; Zheng et al. 2018). In planta expression is a very promising technology due to costs limitation, easy scale-up, absence of risk contamination by human pathogens and possibility of post-translational modifications (Merlin et al. 2014; Moustafa et al. 2016; Tschofen et al. 2016). To date, the production of sweet proteins in transgenic plants has yielded expression levels ranging from 1 to 17% of total soluble proteins (TSP) (Peñarrubia et al. 1992; Lamphear et al. 2005; Sun et al. 2006; Hirai et al. 2011; Hiwasa-Tanase et al. 2012; Pham et al. 2012). In the case of transgenic lettuce and tomato producing the taste-modifying protein miraculin, the nuclear transformation system has shown some limitations, such as gene silencing and the need to improve scale-up and purification processes, to reduce their final production costs (Sun et al. 2006; Hiwasa-Tanase et al. 2012). In the last decades, it has been demonstrated that, among plant-based technologies, chloroplast transformation has many attractive advantages, such as site-specific integration, transgene containment due to maternal inheritance in many crops and high expression levels, up to 70% TSP (Oey et al. 2009; Lentz et al. 2010; Boyhan and Daniell 2011; Castiglia et al. 2016). Nonetheless, previous attempts to express MNEI variants in transplastomic tobacco plants led to low accumulation levels, which, in turn, prevented adequate purification and no evidence of the sweetness of the plant recombinant product was provided (Roh et al. 2006; Lee et al. 2012). The aim of our work was to develop an efficient, stable and low-cost production system in tobacco chloroplasts for three mutant proteins derived from MNEI, namely the hyper-sweet variant Y65R-MNEI, the highly stable E23Q-MNEI, and the “sweeter and stronger” variant E23Q, C41S, Q28K, Y65R-MNEI, dubbed Mut3 (Esposito et al. 2006; Leone et al. 2016; Leone and Picone 2016). Furthermore, we describe a strategy for the efficient purification of the plastid-based recombinant proteins and their full biochemical and functional characterization, which allowed us to compare their properties with those of their bacterial counterparts.
Materials and methods
Plant material
Plants of Nicotiana tabacum L. cv. Petite Havana were grown in sterile conditions on hormone-free medium containing MS salts and B5 vitamins (Duchefa, The Netherlands), 30 g/l sucrose and 8 g/l agar, pH 5.6, with a 16 h photoperiod of 100 µmol photons m−2 s−1 and 8 h dark at 24 °C. Seeds derived from transplastomic plants transformed with genes encoding MNEI mutants (DC and SA series) or with the empty control vector (PRV) and from wild type plants were sown in vitro (in the presence of 500 mg/l of spectinomycin or streptomycin) in the above described condition to confirm homoplasmy, or in soil to analyze plant phenotype.
Chloroplast transformation vectors
pDC vectors were constructed by replacing the neo coding gene in plasmids pHK30 and pHK40 (Kuroda and Maliga 2001a, b) or the L1 coding region in plasmid pPL66 (Lenzi et al. 2008), whereas SA vectors were constructed by replacing the neo coding gene in plasmid pHK40. All plasmids were linearized with NheI and XbaI restriction enzymes. The Y65R-MNEI coding sequence, a variant of the single chain monellin (MNEI) containing the Y65R mutation synthesized by Polymerase Chain Reaction mutagenesis, was amplified from pET22b-Y65R vector (Esposito et al. 2006) to introduce 5′ NheI and 3′ XbaI restriction sites and cloned in pHK40, pHK30 and pPL66 to develop pDC30, pDC31 and pDC32 vectors, respectively. The synthetic MNEI genes, encoding for MNEI variants containing E23Q, Q28K, C41S, Y65R (Mut3) and E23Q mutations, generated using the E. coli codon usage (Leone et al. 2016; Leone and Picone 2016), were amplified from pET22b(+)-Mut3 or -E23Q-MNEI to introduce 5′ NheI and 3′ XbaI restriction sites and cloned in pHK40 to develop pSA1 and pSA11 vectors, respectively. The expression cassettes of pDC30, pSA1 and pSA11 vectors contain the rrn promoter, the 5′ untranslated region (5′-UTR) of E. coli phage T7 gene 10 and the plastid rbcL gene 3′-UTR, whereas pDC31 and pDC32 contain the rrn promoter fused with the 5′ translation control region (5′-TCR), that includes the 5′-UTR and 42 N-terminal nucleotides of the atpB or rbcL open reading frames, respectively, and the plastid rbcL gene 3′-UTR. The insertion of the NheI restriction site downstream the 5′-UTR or 5′TCR introduced an additional translated sequence at the N-termini of all MNEI mutants, corresponding to the MAS tripeptide.
Stable chloroplast transformation
DNAs from pDC and pSA plastid expression vectors were extracted by Plasmid Maxi Kit (Qiagen, Germany) and used for delivery in tobacco leaf tissue. Biolistic experiments, in vitro regeneration and selection of shoots were carried out according to the protocol described in Scotti and Cardi (2012). Homoplasmic transplastomic lines were rooted and propagated on medium containing MS salts with B5 vitamins, 30 g/1 sucrose, 0.1 mg/l NAA, 80 g/l agar and with 500 mg/l of spectinomycin under controlled conditions, and subsequently transferred to soil in a growth chamber (14 h light, 200 µmol photons m−2 s−1, at 25 °C, and 10 h dark at 20 °C) for seeds production.
Southern blot analysis
Total DNA was isolated from leaves of transplastomic and control plants with the DNeasy® Plant Mini kit (Qiagen). DNA (1–2 μg) was digested with BamHI and analyzed according to the procedure described in Scotti and Cardi (2012). The probe corresponding to rrn16–rps12 plastid recombination site, labeled with digoxigenin-11 dUTP (Roche Applied Science, Germany), was obtained by PCR. Chemiluminescent signal was measured using a ChemiDoc™ XRS + and images were analyzed using the Image Lab™ Software (Bio-Rad).
Protein extraction, purification and immunoblot analysis
Leaf total soluble proteins were extracted according to a modified Petersen and Bock (2011) protocol. Extraction buffer contained 50 mM sodium acetate, pH 5.2, 10 mM potassium acetate, 5 mM magnesium acetate, 1 mM EDTA, 1 mM DTT 1 mM PMSF, 1 × complete proteinase inhibitor (Sigma, Missouri, USA) and 1% 2-mercaptoethanol. Samples were resolved by SDS-PAGE in 18% gels that were either stained with Coomassie Brilliant Blue R-250 (Sigma) or blotted onto nitrocellulose membrane (Hybond ECL, GE Healthcare). Membranes were incubated with anti-Y65R-MNEI antibody (1:200, ON at 4 °C) obtained from Primmbiotech through rabbit immunization with E. coli-derived Y65R-MNEI, and subsequently with a HRP-conjugated anti-rabbit antibody (1:60,000, 1 h at RT). The expression levels were quantified using a dilution series of purified E. coli-derived Y65R-MNEI protein and Bio-Rad Image Lab software.
Crude extracts of transplastomic plants were enriched by two subsequent thermal treatments of 15 min each at 60 °C. Extracts from SA1 and SA11 transplastomic plants were also treated at 70 °C or with a unique treatment of 20 min at 60 °C. Heat treated samples, clarified by centrifugation at 20,000g, for 30 min at 4 °C, were dialyzed against 50 mM acetic acid and then applied to a 5 ml Macro-prep High-S cation exchange column (Bio-Rad) in 50 mM sodium acetate, pH 5.5. Elution was performed with a linear gradient from 0 to 500 mM sodium chloride in the same buffer over 20 CV at 2 ml/min. Fractions containing the protein of interest were pooled, desalted and lyophilized. Finally, samples were purified on a Superdex75 HR 10/300 (GE Life Sciences) in 50 mM acetic acid with 150 mM NaCl. Protein containing fractions were desalted and lyophilized. Desalting steps were performed on a 15 ml Sephadex G-25 column in 50 mM acetic acid. Protein purity was assessed by mass spectrometry (see below).
Mass spectrometry
Positive Linear and Reflectron MALDI spectra were recorded on a 5800 MALDI-ToF/ToF (Sciex, Framingham, MA) as described by De Lorenzo et al. (2005). Mass calibration was performed using external peptide standards from Sciex. MS/MS analyses were performed using 1 kV collision energy with air as CID gas.
Circular dichroism
Circular dichroism (CD) spectra were recorded in 20 mM phosphate buffer, pH 5.1 on a Jasco J-715 spectropolarimeter according to previously published protocols (Leone et al. 2015). A concentration of 0.2 mg/mL protein was used for each sample. Thermal denaturation experiments were recorded and analyzed as previously described (Leone et al. 2016).
Taste evaluation
Sweetness threshold was evaluated by triangle test on a panel of 5 subjects (2 males and 3 females), as previously described (Esposito et al. 2006; Leone et al. 2016; Masuda et al. 2016). Solutions of the proteins produced in E. coli were used as positive control and water as negative control. Three paper cups, one containing 5 mL of protein solution and the others containing 5 mL of mineral water, were given to the panelists, who were asked to indicate which cup had the taste-eliciting solution and to rate the taste from 0 (no taste) to 5. A value of 1 indicated the perception of a taste, 2 meant the taste was recognized as sweet. Sample solutions were provided from the lowest (0.15 mg/l) to the highest (2.4 mg/l) concentration. Data are presented as mean ± SEM. All methods were carried out in accordance with relevant guidelines and regulations. All experimental protocols were approved by the Ethics Committee of the University of Naples Federico II. Written informed consent was obtained from all study participants.
Results
Vector construction and production of MNEI transplastomic plants
To obtain high-level expression of MNEI mutants in tobacco plastome, we developed five plastid transformation vectors. All constructs had the same constitutive ribosomal RNA operon promoter (Prrn) and the plastid rbcL gene 3′-UTR (TrbcL) (Fig. 1). Three out of five constructs (pDC series) contained the Y65R-MNEI coding sequence (Esposito et al. 2006), with different 5′ regulatory sequence. In pDC30 vector, the transgene expression was under control of the gene 10 5′-UTR (T7g10) from phage T7 (Kuroda and Maliga 2001a), whilst in pDC31 and pDC32 the expression of the Y65R-MNEI gene was controlled by the 5′ translation control region (5′-TCR) that includes the 5′-UTR and 42 N-terminal nucleotides (DB) of the atpB or rbcL open reading frames (Kuroda and Maliga 2001b), respectively (Fig. 1). In the pSA vector series, the expression of E. coli-optimized Mut3 and E23Q-MNEI transgenes (Leone et al. 2016; Leone and Picone 2016) relied on the T7g10 5′-UTR (Fig. 1). All plastid transformation vectors developed here target exogenous DNA to the trnV-rps12/7 region of the tobacco plastid genome and contain aminoglycoside 3′ adenylyltransferase (aadA) gene as selectable marker.

Plastid vectors and expression cassettes used in transformation experiments containing the transgene encoding different variants of MNEI. For each vector, regulatory sequences, protein accumulation level and phenotype in corresponding transplastomic plants are indicated
Biolistic transformation of tobacco plants produced several primary spectinomycin-resistant shoots for each vector that were subjected to one regeneration round on medium containing spectinomycin to isolate homoplasmic lines. Correct integration of the transgenes in the plastid genome of regenerated transplastomic plants was detected by Southern blot analysis using the targeting sequence as probe (Fig. 2). For all recombinant vectors homoplasmic plants were identified by detection of a 4.9 kb containing the coding sequences of the different MNEI mutants, whilst a 3.3 kb fragment was detected for wild type plants (Fig. 2b). The homoplasmy was also confirmed by lack of phenotypic segregation in inheritance assay (Fig. S1). In Fig. 2b, heteroplasmic lines are also shown, marked with an asterisk, containing both wild type and transformed plastomes. These lines were subjected to an additional regeneration round on medium containing spectinomycin.

Identification of independently generated transplastomic tobacco lines per each construct. Schematic representation of the rps12/7-trnV targeting region in the plastid genome (Nt-ptDNA) and maps of transformed (Nt-DC and Nt-SA series) plastid genome regions involved in transgene integration (a). Southern blot analyses of plastid transformants to select homoplasmic lines per each construct. Heteroplasmic (marked with an asterisk and containing both wild type and transformed plastomes) or untransformed lines (marked with two asterisks) were also shown (b). goi = gene of interest; P = promoter; L = 5′-UTR or leader sequence; DB = downstream box corresponding to 42 N-terminal nucleotides of the coding region of either atpB or rbcL tobacco gene
Plant phenotypes and expression of MNEI protein mutants in transplastomic plants
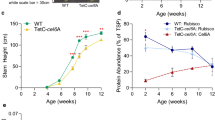
The accumulation of MNEI mutants in tobacco chloroplasts generally did not interfere with plant growth (Figs. 3a, 4a). Transplastomic plants exhibited a phenotype comparable with control plants (wild type and transformed with empty vector), except DC31 transplastomic plant that showed a slight delay in growth (Fig. 3a). All transplastomic plants were able to reach maturity, flower and produce viable seeds by selfing.

Transplastomic plants (T1 generation) producing the Y65R mutant of MNEI sweet protein. Phenotype of control (PRV and PH, transformed with the empty vector and wild type, respectively) and transplastomic plants grown under photoautotrophic conditions in soil (a). Detection of Y65R-MNEI protein accumulated in transplastomic DC plants by western blot analysis. Different amounts of Y65R standard protein and leaf protein extracts were tested. Each transplastomic line shows a 12 kDa protein band corresponding in size to the foreign protein (b). Y65R = E. coli-derived Y65R-MNEI standard protein

Transplastomic plants (T1 generation) producing the Mut3 (SA1) and E23Q-MNEI (SA11) variants of MNEI sweet protein. Comparison of plant growth between control plants (PRV and PH, transformed with the empty vector and wild type, respectively) and transplastomic plants under photoautotrophic conditions in soil (a). Detection of MNEI protein variants accumulated in transplastomic SA plants by western blot analysis. Different amounts of Y65R standard protein and leaf protein extracts were tested. Each transplastomic line shows a 12 kDa protein band corresponding in size to the foreign protein (b). Y65R = E. coli-derived Y65R-MNEI standard protein
Western blot analysis, using E. coli-derived Y65R-MNEI (about 12 kDa) as reference protein, revealed different yields of Y65R-MNEI within the DC series of transplastomic plants. In particular, a 5 and 1.5% TSP yield, corresponding to ~ 160 and 65 µg/g of Fresh Weight (FW), was measured for DC30 and DC31, respectively (Figs. 1, 3b). DC32 plants accumulated recombinant Y65R-MNEI at levels undetectable by Western blot. Based on phenotype and protein yield results, DC30 transplastomic plants were selected for subsequent analyses. Immunoblot analysis performed on crude extracts of SA1 and SA11 plant leaves indicated that the expression of Mut3 and E23Q reached up to 20 and 50% of TSP, corresponding to ~ 0.50 or 1 mg/g FW, respectively (Figs. 1, 4b). It is well-known that biosynthesis of recombinant proteins could vary in leaves according to their developmental stage. Therefore, the accumulation profile of MNEI variants as function of leaf age was also investigated in subsequent generations of transplastomic plants (Fig. S2a). DC30 transplastomic plants showed variable accumulation of Y65R-MNEI in plant leaves, with the highest protein yield detected in old leaves (Fig. S2b), whilst no significant variation in protein accumulation was highlighted for Mut3 (SA1 plants) and E23Q-MNEI (SA11 plants) (Fig. S2c).
Purification of recombinant MNEI protein mutants from transplastomic plants
Leaf crude extracts of DC30, SA1 and SA11 transplastomic plants were subjected to different thermal treatments, to test the possibility of enriching them in MNEI mutant proteins, taking advantage of their high thermal stability. For DC30 plants, the treatment at 60 °C for 15 min did not reveal a significant enrichment in Y65R-MNEI protein (Fig. S3a), in fact, the T1 and T2 fractions showed the same protein profile of the crude extract.
On the contrary, protein samples of SA1 and SA11 plants heated to 60 or 70 °C produced a better enrichment in the proteins of interest (Fig. S3b, c), being that most of the plant proteins precipitated in the P1 fraction as showed in Coomassie stained SDS-PAGE. Because no significant difference was evident between the thermal treatment at 60 or 70 °C for both Mut3 and E23Q (Figs. S3b, c), we chose the thermal treatment at 60 °C for 20 min for subsequent downstream processes and biochemical characterization.
Despite the good enrichment indicated by SDS-PAGE, analytical Size Exclusion FPLC and Circular Dichroism (CD) spectroscopy showed abnormal patterns also in the case of Mut3 and E23Q-MNEI, due to the presence of protein and non-protein contaminants. Therefore, thermally treated extracts were subjected to a double round of chromatography using first a strong cationic exchange resin followed by size exclusion chromatography, which yielded each mutant with a purity of at least 99% (see materials and methods section for details).
Structure characterization of the purified proteins
Pure protein samples from E. coli and from tobacco leaves were compared by MALDI-ToF analysis (Fig S4). All proteins were correctly expressed in the plant system, as confirmed by the MS data summarized in Table 1, showing, for each species, an m/z consistent with the theoretical molecular weight. All MNEI variants produced in plants gave, as expected, peaks with higher molecular weight than the homologous bacterial counterparts, due to the introduction of the tripeptide MAS at the N-terminus of the proteins by the cloning procedure. Moreover, the observed m/z values indicated that quantitative cleavage of the N-terminal methionine had occurred for all plant constructs, whilst it was retained in all bacterial proteins. Hence, the plant proteins ultimately differed from their bacterial homologues by two amino acids, i.e., the AS dipeptide, at the N-terminus. MALDI mapping after trypsin digestion and MS/MS fragmentation experiments on selected ion peaks confirmed the correct expression of the plastid-based proteins and a 100% sequence identity. As an example, Fig. 5 shows the fragmentation pattern of the ion at m/z 2206.02, corresponding to the N-terminal peptide of the plastid-derived proteins. MALDI-ToF mapping results are summarized in Table S1.

MS/MS fragmentation spectrum of the peptide with m/z 2206.02, corresponding to the N-terminal fragment of all plant-derived constructs. The peptide sequence deduced from the observed fragmentation pattern, according to the b and y ion series, was showed
The similarity of the CD spectra recorded on the proteins purified from E. coli and from tobacco leaves confirmed that the plastid-based proteins assume the same fold of the reference proteins (Fig. S5). CD spectroscopy was also used to evaluate the proteins’ stability toward thermal denaturation. Figure 6 shows that the denaturation profiles of all the proteins from transplastomic plants are comparable to their bacterial homologues.

Circular Dichroism (CD) unfolding curves at pH 5.1 of Y65R-MNEI, Mut3 and E23Q-MNEI produced in tobacco chloroplast (green) or in E. coli (red). All mutants show comparable stability independently from the production host
Functional characterization of the purified proteins
The sweetness threshold of all protein samples was determined by triangle test (Esposito et al. 2006; Masuda et al. 2016; Leone et al. 2016). All plant-derived proteins revealed a sweetness threshold of 1.08 ± 0.04 mg/l, 1.64 ± 0.04 mg/l and 0.40 ± 0.02 mg/l for Y65R-MNEI, E23Q-MNEI and Mut3, respectively. In comparison, proteins produced in E. coli had a threshold of 0.72 ± 0.02 mg/l, 0.96 ± 0.02 mg/l and 0.25 ± 0.03 mg/l, respectively). Hence, all plant-based constructs were marginally less sweet than their bacterial counterparts (Fig. 7).

Sweetness thresholds of the different MNEI mutants produced in plants (green) or in E. coli (red). All proteins possess comparable sweetness to their bacterial homologous
Discussion
Over the past decades, a wide range of heterologous expression systems has been developed for the production of recombinant proteins with industrial applications (Demain and Vaishnav, 2009; Sanchez-Garcia et al. 2016; Zhou et al. 2018). Among them, plants have been proposed as an affordable, safe and easily scalable production system, characterized, in recent years, by exciting biotechnological progresses (Lomonossoff and D’Aoust 2016; Moustafa et al. 2016; Tschofen et al. 2016). In particular, the extraordinarily high expression levels obtained for proteins produced by plastome transformation (Oey et al. 2009; Lentz et al. 2010; Boyhan and Daniell 2011; Castiglia et al. 2016) has opened new perspectives to plant biotechnologists. In this work, we exploited the possibility to use tobacco chloroplasts as a stable and cheap production platform for high expression levels of MNEI mutant proteins, which could become alternative sweeteners, employed also by people suffering of chronic diseases (e.g., obesity, diabetes and hyperlipemia). After its start in the 70s, research on sweet proteins has in fact had a recent comeback, due to the need of substituting sucrose in the diet to stem the spreading of these pathologies, both in western world and developing countries. The use of sweet proteins as sugar replacers has been traditionally limited by various factors: the difficulty of farming the tropical plants from which they could be extracted; the intrinsic structural lability of the proteins upon exposure to harsh environmental factors and, in some cases, the lack of thorough evaluations of the effects of their consumption on human health. To date, in fact, only thaumatin has been approved by food agencies for human consumption, in consequence of its recognized lack of toxicity (Higginbotham et al. 1983). Recent experimental work, though, indicates the absence of mutagenetic effects and environmental toxicity for MNEI (Rega et al. 2017), which bodes well for future studies in this direction and for the possibility of extending the safe status also to monellin derivatives. We focused on the hyper-sweet Y65R-MNEI, the highly stable E23Q-MNEI, and the “sweeter and stronger” Mut3 (Esposito et al. 2006; Leone et al. 2016; Leone and Picone 2016). The production of constructs with improved characteristics, which overcome the intrinsic limits of natural wild type monellin, is in fact one of the most important benefits of the recombinant production of these proteins. All MNEI mutants were stably expressed in tobacco chloroplasts, with protein yields ranging from 5% to more than 50% TSP for Y65R-MNEI and E23Q-MNEI, respectively. These high expression levels had no negative effect on plant phenotype. It is well-known that many factors, such as gene regulatory sequences (promoter, 5′-UTR or TCR, 3′-UTR), codon usage, intrinsic properties of recombinant proteins, plant tissue and species, can affect the protein accumulation level in transgenic chloroplasts (Scotti et al. 2009; Apel et al. 2010; Castiglia et al. 2016). Since in this work, the maximum yield for each MNEI mutant was obtained using the 5′-UTR of gene 10 of the phage T7 as regulatory sequence, the variable expression level observed between the variants tested could be due to both codon usage and intrinsic property of recombinant proteins. In fact, the highest yields were achieved with the E. coli-optimized Mut3 (20% TSP) and E23Q-MNEI (50% TSP) transgenes. These two mutant proteins also showed, compared to Y65R-MNEI, a more stable and constant accumulation level in leaves at different developmental stage, and a good enrichment by thermal treatment of leaf extracts. Compared to previous studies on the expression of MNEI mutants in transplastomic plants using different 5′-UTR as regulatory sequence (Roh et al. 2006; Lee et al. 2012), the protein yields here reported represent the highest accumulation levels for the production of MNEI mutants in plants so far. In particular, the highest yield obtained in the tobacco plastome (5% TSP) with MNEI-E24W mutant (Lee et al. 2012) is comparable to the accumulation level we achieved with the Y65R-MNEI protein, the mutant characterized by the lowest protein yield. Furthermore, the plastid-derived MNEI-E24W and other mutants were not characterized from biochemical or functional point of view, being these analyses performed on E. coli-derived protein mutants (Lee et al. 2012). In this study, we report, for the first time, the development of a purification procedure for MNEI protein mutants produced in transplastomic plants that allowed for a 50% recovery of pure protein (with 99% of purity). Purification procedure is still a crucial and limiting downstream process for recombinant proteins as demonstrated by the poor recovery (400 µg of miraculin from 100 g of transgenic tomato leaves) achieved by Sun et al. (2007) through a purification protocol based on ammonium sulfate precipitation and three rounds of chromatography. Based on the accumulation level and the percentage of pure protein recovery we could estimate a lab-scale production up to 25–30 mg of MNEI mutant protein/plant, notably, without the introduction of affinity tags. Usually, the use of an affinity tag permits the development of a faster purification protocol based on a single chromatography step but for such proteins this is an important issue, because even small modifications in the sequence of sweet proteins could dramatically affect their function. In fact, all sweet proteins own their peculiar taste profile to the possibility of binding to the dimeric sweet taste receptor T1R2-T1R3 (Cui et al. 2006). This interaction occurs according to the so-called “wedge model”, which involves several surface residues of the protein binding to a cleft encompassing both subunit of the receptor (Morini et al. 2005; Temussi 2011a and b) and is only possible when the protein is correctly folded and the proper amino acids pattern is displayed on the protein surface. The purity and identity of plastid-based recombinant proteins was demonstrated by MALDI-ToF analysis, which showed peaks with higher molecular weight than their bacterial counterparts due to the insertion, for cloning purposes, of the tripeptide MAS at the N-terminus of the proteins. This analysis also showed that the start methionine was correctly cleaved in all plastid-derived proteins, a mechanism in many cases related to the correct functionality of the synthesized proteins (Giglione et al. 2003). By contrast, the proteins produced in E. coli retained the methionine, a phenomenon sometimes associated to protein overproduction, which, in our case, fortunately did not affect the proteins’ taste (Giglione et al. 2004). No other sequence differences were revealed by MALDI-ToF and MS/MS analyses. Additional biochemical analyses confirmed the high similarity, in terms of structure, stability and function, of the proteins produced in plant chloroplasts and bacteria. The small sequence differences, localized at the N-terminus, might slightly affect the interaction between the protein and the sweet taste receptor and be responsible of the marginally lower sweetness of plastid-based MNEI mutants compared to their bacterial homologues. Nevertheless, based on results of E. coli-derived MNEI protein that showed a Recognition Threshold (RT) 3000 times lower than sucrose (Di Monaco et al. 2013), we could assume that all plastid-derived proteins, even if they showed a slightly lower sweetness compared to their bacterial counterparts, still exhibited a remarkable sweetness, and hence they can be considered promising candidates as high intensity hypocaloric sweeteners. The high protein yields obtained in this study confirm that plastid transformation technology is a realistic, safe, stable (in all plant generation tested), and cost-effective production platform for low-calorie sweeteners, because once a homoplasmic transplastomic plant is achieved and seeds are collected, they can be grown and scaled up according to the requirements without particular equipment either in academic research laboratory or at industrial scale. On the contrary, fermenter-based systems require equipment and growth media, which affects the production costs (Waheed et al. 2015). No significant difference in term of costs was observed between plant- and fermenter-based production systems for downstream processing. Further work will be necessary to complete the direct comparison of the properties of plastid-based MNEI mutants and sucrose.
Author contribution statement
SG and NS conceived and designed plant research. SL and DP conceived and designed biochemical research. DC, RT and LS performed all experiments on plants. SL, JF, CM and AC performed all biochemical experiments. All authors contributed to data analysis and writing the manuscript.
References
Apel W, Schulze WX, Bock R (2010) Identification of protein stability determinants in chloroplasts. Plant J 63:636–650
Boyhan D, Daniell H (2011) Low-cost production of proinsulin in tobacco and lettuce chloroplasts for injectable or oral delivery of functional insulin and C-peptide. Plant Biotechnol J 9:585–598
Cai C, Li L, Lu N, Zheng W, Yang L, Liu B (2016) Expression of a high sweetness and heat-resistant mutant of sweet-tasting protein, monellin, in Pichia pastoris with a constitutive GAPDH promoter and modified N-terminus. Biotechnol Lett 38:1941–1946
Castiglia D, Sannino L, Marcolongo L, Ionata E, Tamburino R, De Stradis A, Cobucci-Ponzano B, Moracci M, La Cara F, Scotti N (2016) High-level expression of thermostable cellulolytic enzymes in tobacco transplastomic plants and their use in hydrolysis of an industrially pretreated Arundo donax L. biomass. Biotechnol Biofuels 9:154
Chen Z, Heng C, Li Z, Liang X, Xinchen S (2007) Expression and secretion of a single-chain sweet protein monellin in Bacillus subtilis by sacB promoter and signal peptide. Appl Microbiol Biotechnol 73:1377–1381
Chen Z, Li Z, Yu N, Yan L (2011) Expression and secretion of a single-chain sweet protein, monellin, in Saccharomyces cerevisiae by an α-factor signal peptide. Biotechnol Lett 33:721–725
Cui M, Jiang P, Maillet E, Max M, Margolskee RF, Osman R (2006) The heterodimeric sweet taste receptor has multiple potential ligand binding sites. Curr Pharm Des 12:4591–4600
De Lorenzo C, Cozzolino R, Carpentieri A, Pucci P, Laccetti P, D’Alessio G (2005) Biological properties of a human compact anti-ErbB2 antibody. Carcinogenesis 26:1890–1895
Demain AL, Vaishnav P (2009) Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv 27:297–306
Di Monaco R, Miele NA, Picone D, Masi P, Cavella S (2013) Taste detection and recognition thresholds of the modified monellin sweetener: MNEI. J Sens Stud 28:25–33
Di Monaco R, Miele NA, Volpe S, Picone D, Cavella S (2014) Temporal sweetness profile of MNEI and comparison with commercial sweeteners. J Sens Stud 29:385–394
Esposito V, Gallucci R, Picone D, Saviano G, Tancredi T, Temussi PA (2006) The importance of electrostatic potential in the interaction of sweet proteins with the sweet taste receptor. J Mol Biol 360:448–456
Giglione C, Vallon O, Meinnel T (2003) Control of protein life-span by N-terminal methionine excision. EMBO J 22:13–23
Giglione C, Boularot A, Meinnel T (2004) Protein N-terminal methionine excision. Cell Mol Life Sci 61:1455–1474
Higginbotham JD, Snodin DJ, Eaton KK, Daniel JW (1983) Safety evaluation of thaumatin (Talin protein). Food Chem Toxicol Int J Publ Br Ind Biol Res Assoc 21:815–823
Hirai T, Kurokawa N, Duhita N, Hiwasa-Tanase K, Kato K, Kato K, Ezura H (2011) The HSP terminator of Arabidopsis thaliana induces a high level of miraculin accumulation in transgenic tomatoes. J Agric Food Chem 59:9942–9949
Hiwasa-Tanase K, Hirai T, Kato K, Duhita N, Ezura H (2012) From miracle fruit to transgenic tomato: mass production of the taste-modifying protein miraculin in transgenic plants. Plant Cell Rep 31:513–525
Hobbs JR, Munger SD, Conn GL (2007) Monellin (MNEI) at 1.15 Å resolution. Acta Crystallogr, Sect F Struct Biol Cryst Commun 63:162–167
Inglett GE, May JF (1969) Serendipity berries–source of a new intense sweetener. J Food Sci 34:408–411
Kant R (2005) Sweet proteins-Potential replacement for artificial low calorie sweeteners. Nutr J 4:5
Kaul T, Reddy CS, Pandey S (2018) Transgenics with monellin. In: Mérillon JM, Ramawat KG (eds) Sweeteners. Pharmacology, biotechnology and applications. Springer, Cham, pp 211–222
Kim IH, Lim KJ (1996) Large-scale purification of recombinant monellin from yeast. J Ferment Bioeng 82:180–182
Kim SH, Kang CH, Kim R, Cho JM, Lee YB, Lee TK (1989) Redesigning a sweet protein: increased stability and renaturability. Protein Eng Des Sel 2:571–575
Kuroda H, Maliga P (2001a) Complementarity of the 16S rRNA penultimate stem with sequences downstream of the AUG destabilizes the plastid mRNAs. Nucleic Acids Res 29:970–975
Kuroda H, Maliga P (2001b) Sequences downstream of the translation initiation codon are important determinants of translation efficiency in chloroplasts. Plant Physiol 125:430–436
Lamphear BJ, Barker DK, Brooks CA, Delaney DE, Lane JR, Beifuss K, Love R, Thompson K, Mayor J, Clough R, Harkey R, Poage M, Drees C, Horn ME, Streatfield SJ, Nikolov Z, Woodard SL, Hood EE, Jilka JM, Howard JA (2005) Expression of the sweet protein brazzein in maize for production of a new commercial sweetener. Plant Biotechnol J 3:103–114
Lee S-B, Kim Y, Lee J, Oh K-J, Byun M-O, Jeong M-J, Bae S-C (2012) Stable expression of the sweet protein monellin variant MNEI in tobacco chloroplasts. Plant Biotechnol Rep 6:285–295
Lentz EM, Segretin ME, Morgenfeld MM, Wirth SA, Dus Santos MJ, Mozgovoj MV, Wigdorovitz A, Bravo-Almonacid FF (2010) High expression level of a foot and mouth disease virus epitope in tobacco transplastomic plants. Planta 231:387–395
Lenzi P, Scotti N, Alagna F, Tornesello ML, Pompa A, Vitale A, De Stradis A, Monti L, Grillo S, Buonaguro FM, Maliga P, Cardi T (2008) Translational fusion of chloroplast-expressed human papillomavirus type 16 L1 capsid protein enhances antigen accumulation in transplastomic tobacco. Transgenic Res 17:1091–1102
Leone S, Sannino F, Tutino ML, Parrilli E, Picone D (2015) Acetate: friend or foe? Efficient production of a sweet protein in Escherichia coli BL21 using acetate as a carbon source. Microb Cell Fact 14:106
Leone S, Pica A, Merlino A, Sannino F, Temussi PA, Picone D (2016) Sweeter and stronger: enhancing sweetness and stability of the single chain monellin MNEI through molecular design. Sci Rep 6:34045
Leone S, Picone D (2016) Molecular dynamics driven design of pH-stabilized mutants of MNEI, a sweet protein. PLoS One 11:e0158372
Liu Q, Li L, Yang L, Liu T, Cai C, Liu B (2016) Modification of the sweetness and stability of sweet-tasting protein monellin by gene mutation and protein engineering. Biomed Res Int. https://doi.org/10.1155/2016/3647173
Lomonossoff GP, D’Aoust MA (2016) Plant-produced biopharmaceuticals: a case of technical developments driving clinical deployment. Science 16:1237–1240
Masuda T, Kitabatake N (2006) Developments in biotechnological production of sweet proteins. J Biosci Bioeng 102:375–389
Masuda T, Ohta K, Ojiro N, Murata K, Mikami B, Tani F, Temussi PA, Kitabatake N (2016) A hypersweet protein: removal of the specific negative charge at Asp21 enhances thaumatin sweetness. Sci Rep 6:20255
Merlin M, Gecchele E, Capaldi S, Pezzotti M, Avesani L (2014) Comparative evaluation of recombinant protein production in different biofactories: the green perspective. Biomed Res Int. https://doi.org/10.1155/2014/136419
Miele NA, Cabisidan EK, Blaiotta G, Leone S, Masi P, Monaco RD, Cavella S (2017a) Rheological and sensory performance of a protein-based sweetener (MNEI), sucrose, and aspartame in yogurt. J Dairy Sci 100:9539–9550
Miele NA, Di Monaco R, Dell’Amura F, Rega MF, Picone D, Cavella S (2017b) A preliminary study on the application of natural sweet proteins in agar-based gels. J Texture Stud 48:103–113
Morini G, Bassoli A, Temussi PA (2005) From small sweeteners to sweet proteins: anatomy of the binding sites of the human T1R2_T1R3 receptor. J Med Chem 48:5520–5529
Morris JA, Cagan RH (1972) Purification of monellin, the sweet principle of Dioscoreophyllum cumminsii. Biochim Biophys Acta 261:114–122
Moustafa K, Makhzoum A, Trémouillaux-Guiller J (2016) Molecular farming on rescue of pharma industry for next generations. Crit Rev Biotechnol 36:840–850
Murzin AG (1993) Sweet-tasting protein Monellin is related to the cystatin family of thiol proteinase inhibitors. J Mol Biol 230:689–694
Oey M, Lohse M, Kreikemeyer B, Bock R (2009) Exhaustion of the chloroplast protein synthesis capacity by massive expression of a highly stable protein antibiotic. Plant J 57:436–445
Ogata C, Hatada M, Tomlinson G, Shin W-C, Kim S-H (1987) Crystal structure of the intensely sweet protein monellin. Nature 328:739–742
Peñarrubia L, Kim R, Giovannoni J, Kim S-H, Fischer RL (1992) Production of the sweet protein monellin in transgenic plants. Nat Biotechnol 10:561–564
Petersen K, Bock R (2011) High-level expression of a suite of thermostable cell wall-degrading enzymes from the chloroplast genome. Plant Mol Biol 76:311–321
Pham NB, Schäfer H, Wink M (2012) Production and secretion of recombinant thaumatin in tobacco hairy root cultures. Biotechnol J 7:537–545
Pica A, Leone S, Di Girolamo R, Donnarumma F, Emendato A, Rega MF, Merlino A, Picone D (2018) pH driven fibrillar aggregation of the super-sweet protein Y65R-MNEI: a step-by-step structural analysis. Biochim Biophys Acta 1862:808–815
Picone D, Temussi PA (2012) Dissimilar sweet proteins from plants: oddities or normal components? Plant Sci 195:135–142
Reddy CS, Vijayalakshmi M, Kaul T, Islam T, Reddy MK (2015) Improving flavour and quality of tomatoes by expression of synthetic gene encoding sweet protein monellin. Mol Biotechnol 57:448–453
Rega MF, Di Monaco R, Leone S, Donnarumma F, Spadaccini R, Cavella S, Picone D (2015) Design of sweet protein based sweeteners: hints from structure-function relationships. Food Chem 173:1179–1186
Rega MF, Siciliano A, Gesuele R, Lofrano G, Carpentieri A, Picone D, Guida M (2017) Ecotoxicological survey of MNEI and Y65R-MNEI proteins as new potential high-intensity sweeteners. Environ Sci Pollut Res 24:9734–9740
Roh KH, Shin K-S, Lee Y-H, Seo S-C, Park H-G, Daniell H, Lee S-B (2006) Accumulation of sweet protein monellin is regulated by the psbA 5′UTR in tobacco chloroplasts. J Plant Biol 49:34–43
Sanchez-Garcia L, Martín L, Mangues R, Ferrer-Miralles N, Vázquez E, Villaverde A (2016) Recombinant pharmaceuticals from microbial cells: a 2015 update. Microb Cell Fact 15:33
Scotti N, Alagna F, Ferraiolo E, Formisano G, Sannino L, Buonaguro L, De Stradis A, Vitale A, Monti L, Grillo S, Buonaguro FM, Cardi T (2009) High-level expression of the HIV-1 Pr55gag polyprotein in transgenic tobacco chloroplasts. Planta 229:1109–1122
Scotti N, Cardi T (2012) Plastid transformation as an expression tool for plant-derived biopharmaceuticals. In: Dunwell JM, Wetten AC (eds) Transgenic plants. Methods and protocols, 2nd edn. Humana Press, New York, pp 451–466
Spadaccini R, Crescenzi O, Tancredi T, De Casamassimi N, Saviano G, Scognamiglio R, DonatoA Di, Temussi PA (2001) Solution structure of a sweet protein: NMR study of MNEI, a single chain monellin. J Mol Biol 305:505–514
Spadaccini R, Leone S, Rega MF, Richter C, Picone D (2016) Influence of pH on the structure and stability of the sweet protein MNEI. FEBS Lett 590:3681–3689
Sun H-J, Cui M, Ma B, Ezura H (2006) Functional expression of the taste-modifying protein, miraculin, in transgenic lettuce. FEBS Lett 580:620–626
Sun H-J, Kataoka H, Yano M, Ezura H (2007) Genetically stable expression of functional miraculin, a new type of alternative sweetener, in transgenic tomato plants. Plant Biotechnol J 5:768–777
Tancredi T, Iijima H, Saviano G, Amodeo P, Temussi PA (1992) Structural determination of the active site of a sweet protein A 1H NMR investigation of pMNEI. FEBS Lett 310:27–30
Tancredi T, Pastore A, Salvadori S, Esposito V, Temussi PA (2004) Interaction of sweet proteins with their receptor. Eur J Biochem 271:2231–2240
Temussi PA (2002) Why are sweet proteins sweet? Interaction of brazzein, monellin and thaumatin with the T1R2-T1R3 receptor. FEBS Lett 526:1–4
Temussi PA (2011a) New insights into the characteristics of sweet and bitter taste receptors. In: Jeon KW (ed) International Review of Cell and Molecular Biology, vol 291. Academic Press, San Diego, pp 191–226
Temussi PA (2011b) Determinants of sweetness in proteins: a topological approach. J Mol Recognit 24:1033–1042
Tschofen M, Knopp D, Hood E, Stöger E (2016) Plant molecular farming: much more than medicines. Annu Rev Anal Chem 9:271–294
van der Wel H, Loeve K (1972) Isolation and characterization of thaumatin I and II, the sweet-tasting proteins from Thaumatococcus daniellii Benth. Eur J Biochem 31:221–225
Waheed MT, Ismail H, Gottschamel J, Mirza B, Lössl AG (2015) Plastids: the green frontiers for vaccine production. Front Plant Sci 6:1005
Zheng W, Yang L, Cai C, Ni J, Liu B (2018) Expression, purification and characterization of a novel double-site mutant of the single-chain sweet-tasting protein monellin (MNEI) with both improved sweetness and stability. Protein Expr Purif 143:52–56
Zhou Y, Lu Z, Wang X, Selvaraj JN, Zhang G (2018) Genetic engineering modification and fermentation optimization for extracellular production of recombinant proteins using Escherichia coli. Appl Microbiol Biotechnol 102:1545–1556
Acknowledgements
This work was partially funded by Fondazione con il Sud, Project 2011-PDR-19, and POR Campania FESR207-2013, Project “Bio Industrial Processes (BIP)”. Technical assistance of Mr. G. Guarino, Ms. S. Arcari and Mr. R. Nocerino (CNR-IBBR, Portici, Italy) with artworks and plant growth is gratefully acknowledged. The authors declare that they have no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Castiglia, D., Leone, S., Tamburino, R. et al. High-level production of single chain monellin mutants with enhanced sweetness and stability in tobacco chloroplasts. Planta 248, 465–476 (2018). https://doi.org/10.1007/s00425-018-2920-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-018-2920-z