
Abstract
Main conclusion
Localization of the RNase RNS2 to the vacuole via a C-terminal targeting signal is essential for its function in rRNA degradation and homeostasis.
RNase T2 ribonucleases are highly conserved enzymes present in the genomes of nearly all eukaryotes and many microorganisms. Their constitutive expression in different tissues and cell types of many organisms suggests a housekeeping role in RNA homeostasis. The Arabidopsis thaliana class II RNase T2, RNS2, is encoded by a single gene and functions in rRNA degradation. Loss of RNS2 results in RNA accumulation and constitutive activation of autophagy, possibly as a compensatory mechanism. While the majority of RNase T2 enzymes is secreted, RNS2 is located within the vacuole and in the endoplasmic reticulum (ER), possibly within ER bodies. As RNS2 has a neutral pH optimum, and the endomembrane organelles are connected by vesicle transport, the site within the endomembrane system at which RNS2 functions is unclear. Here we demonstrate that localization to the vacuole is essential for the physiological function of RNS2. A mutant allele of RNS2, rns2-1, results in production of an active RNS2 RNase but with a mutation that removes a putative C-terminal vacuolar targeting signal. The mutant protein is, therefore, secreted from the cell. This results in a constitutive autophagy phenotype similar to that observed in rns2 null mutants. These findings illustrate that the intracellular retention of RNS2 and localization within the vacuole are critical for its cellular function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The degradation of RNA and the maintenance of RNA homeostasis are critical processes carried out by the activity of RNases. Many routes exist for the breakdown of RNA in cells (Andersen et al. 2008; Balagopal and Parker 2009; Houseley and Tollervey 2009). The RNase T2 family of acidic transferase-type endoribonucleases represents a widely distributed and conserved class of RNases. RNase T2 proteins are either secreted or targeted to membrane-bound compartments of the secretory pathway such as the ER, lysosome, or vacuole (Irie 1999; MacIntosh 2011). RNase T2 enzymes are found in the genomes of almost all eukaryotic organisms so far analyzed, many prokaryotes, and many viruses (Irie 1999; Hillwig et al. 2009, 2011; MacIntosh et al. 2010; Andersen and Collins 2012) but with some exceptions (Condon and Putzer 2002). The evolutionary conservation of these proteins illustrates their critical role in the physiological process of RNA degradation.
In plants, the RNase T2 family has been divided into three classes based on sequence homology, expression patterns, and intron structure (Igic and Kohn 2001). Class III enzymes, the first of the plant RNase T2 protein classes to be functionally characterized, are involved in self-incompatibility mechanisms in the Plantaginaceae, Solanaceae, Scrophulariaceae, and Rosaceae families (Hua et al. 2008; MacIntosh et al. 2010; Meng et al. 2011). Class I enzymes are highly evolutionarily diversified, tissue specific, and can be regulated by biotic and abiotic stress (Bariola et al. 1994; MacIntosh et al. 2010). Class II enzymes are often expressed constitutively throughout the organism and are widely conserved (MacIntosh et al. 2001, 2011; Henneke et al. 2009; Hillwig et al. 2011).
RNS2 (At2g39780) is the sole class II RNase T2 in Arabidopsis thaliana and it functions in RNA homeostasis as a major RNase activity involved in rRNA degradation (Taylor et al. 1993; Hillwig et al. 2011). A null RNS2 knockout mutant, rns2-2, has rRNA with a longer half-life, accumulates rRNA in the vacuole, and has increased basal autophagy under normal growth conditions (Hillwig et al. 2011; Floyd et al. 2015). Autophagy is a macromolecular recycling pathway in which cellular components are transferred into the vacuole for degradation (Floyd et al. 2012; Yang and Bassham 2015); the upregulation of autophagy in rns2-2 mutants is hypothesized to occur as a compensatory mechanism for lack of RNS2 activity (Floyd et al. 2015). Similar roles in the turnover of intracellular rRNA have been proposed for RNS2 homologues in human, zebrafish, and yeast (Henneke et al. 2009; Haud et al. 2011; Huang et al. 2015).
The site of action of RNS2 has remained unclear. Upon overexpression of RNS2 as a cyan fluorescent protein (CFP) fusion protein, fluorescence was observed in ER bodies and in the vacuole, and RNS2 enzymatic activity was detected in both vacuole and ER fractions after subcellular fractionation (Hillwig et al. 2011). The accumulation of rRNA in the vacuole in the rns2-2 null mutant (Floyd et al. 2015) is highly suggestive of a function for RNS2 within this organelle, but as the organelles of the endomembrane system are connected by extensive vesicular traffic, this does not rule out the possibility that RNS2 functions within the ER. In addition, in vitro enzyme assays have indicated that RNS2 has optimal activity at a more neutral pH than that of the vacuole (Hillwig et al. 2011). It is, therefore, unclear if localization of RNS2 to the vacuole is necessary for its biological function in the cell and whether the constitutive autophagy phenotype observed in the rns2-2 null mutant is due to lack of vacuolar rRNA degradation or related to an additional RNS2 function not yet discovered.
Here we take advantage of a mislocalized but catalytically active mutant allele of RNS2, rns2-1, to determine the importance of the sub-cellular localization of this enzyme. Unlike the rns2-2 null mutant, the RNS2-1 protein retains RNase activity. However, rns2-1 plants show the same increased basal autophagy phenotype as seen in rns2-2 null mutants. Sequence and localization analysis of RNS2-1 showed that the vacuolar localization of the protein is disrupted and instead it is secreted from the cell. Moreover, analysis of RNase activity from purified vacuoles showed no difference between the null mutant and the rns2-1 organelles. These findings illustrate that the intracellular retention of RNS2 and localization within the vacuole is necessary for its biological role in rRNA degradation and maintenance of cellular homeostasis.
Materials and methods
Plant growth and Arabidopsis thaliana genotypes
Arabidopsis thaliana T-DNA insertion mutants rns2-2 (Hillwig et al. 2011) and atg9-4 (Floyd et al. 2015) were previously described. The T-DNA insertion mutant rns2-1 (SAIL_872_B10, stock #CS877567) was obtained from the Arabidopsis Biological Resource Center. All mutants have the Columbia-0 genetic background and this accession was used as wild-type (WT) control. To generate double mutants, rns2-1 and atg9-4 mutants were crossed and genomic DNA from F1 and F2 progeny analyzed by PCR using the primers listed in Table 1 to identify homozygous double mutant lines. For protoplast preparation, plants were grown in soil under short day conditions (10 h light/14 h dark) at 22 °C for 6 weeks. For plate-grown seedlings, seeds were surface sterilized in 33% (v/v) bleach, 1% Triton X-100 for 20 min followed by cold treatment for ≥2 days. Plants were grown for 7 days under long day conditions (16 h light/8 h dark) at 22 °C on half-strength Murashige–Skoog (MS) solid medium with vitamins (MSP09; Caisson Labs), 1% sucrose, 2.4 mM MES (pH5.7), and 0.8% (w/v) phytoagar (PTP01; Caisson Labs) as described (Liu et al. 2012).
RT-PCR and in-gel RNase activity analysis
RNA was extracted from 7-day-old seedlings using Trizol reagent (15596018; Invitrogen) and treated with DNase I (18068015; Invitrogen). cDNA was synthesized using SuperScript III reverse transcriptase (18080093; Invitrogen) and oligo dT primer (C1101; Promega). RT-PCR was completed for 28 cycles using the primers listed in Table 1. In-gel RNase activity assays were performed as previously described using purified high-molecular weight torula yeast RNA (R-6625; Sigma) as substrate (Yen and Green 1991; Hillwig et al. 2011).
Cloning and FLAG-RNS2 and FLAG-RNS2-1 constructs
The rns2-1 cDNA was amplified using a 3′-RACE system (18373-019; Invitrogen) with an internal RNS2 primer (Table 1). The 19 amino acid RNS2 N-terminal signal peptide was predicted by SignalP 4.1 (http://www.cbs.dtu.dk/services/SignalP/) (Petersen et al. 2011) and corresponds to the prediction by Taylor et al. (1993). A FLAG tag was engineered between the signal peptide and mature protein by inclusion in the primers for PCR (Table 1). An additional glycine was included prior to the FLAG tag to aid in proper signal peptide cleavage without disruption of the tag. FLAG-containing constructs were assembled using the Clontech In-Fusion HD cloning plus kit (638909; Clontech). FLAG-RNS2-1 and FLAG-RNS2 fusions were inserted into the pCAMBIA1300MCS1 (Sanderfoot et al. 2001) binary vector (Cambia GPO, Canberra, Australia). FLAG-RNS2 was introduced into the rns2-1 mutant and FLAG-RNS2-1 into the rns2-2 mutant by Agrobacterium tumefaciens mediated transformation using the floral dip method (Clough and Bent 1998).
Staining and microscopy
Seven-day-old seedlings were stained with 50 μM monodansylcadaverine (MDC; 30432; Sigma) in phosphate-buffered saline pH 7.4 according to Contento et al. (2005). Fluorescence and differential interference contrast microscopy used a Zeiss Axio Imager A2 upright fluorescent microscope (Carl Zeiss Inc.). Confocal microscopy was performed using a Leica SP5 X MP confocal multiphoton microscope (Leica Microsystems) and HPX PL APO CS 63.0 × 1.40 oil objective with an excitation/emission of 405 nm/430–550 nm for MDC detection. MDC-stained motile puncta in all cells visible in the focal plane were imaged in the late elongation zone and neighboring cells in the differentiation zone while excluding root tips and older differentiation zone cells. Puncta were counted and expressed per frame from 20 root images per genotype for each of at least three biological replicates.
Protoplast transient transformation
Arabidopsis rosette leaf protoplasts from WT, rns2-1 and rns2-2 plants were isolated and transformed with 30 μg of FLAG-RNS2, FLAG-RNS2-1 or GFP-ATG8e (Contento et al. 2005) plasmid DNA according to Sheen (2002). Plasmid DNA was prepared using a GeneElute HP plasmid maxiprep kit (NA0310; Sigma).
For immunoblotting, protoplasts were incubated for 44 h and collected by centrifugation at 68g. Protein from cell and medium fractions was precipitated using TCA, washed with acetone and dissolved in 50 μl and 30 μl 2 × SDS loading buffer [4% (w/v) SDS, 20% (v/v) glycerol, 125 mM Tris–HCl pH 6.8] respectively. Proteins were separated by SDS–polyacrylamide gel electrophoresis and analyzed by immunoblotting using an anti-FLAG antibody (F1804; Sigma).
For GFP-ATG8e microscopy, protoplasts were treated with 1 µM concanamycin A (ConcA) or DMSO as a solvent control and incubated in darkness for 6 h with shaking at 50 rpm, followed by visualization using a Zeiss Axioplan II light microscope equipped with an Axio Cam HRC digital imaging system and 40x objective (Carl Zeiss Inc., Jena, Germany). Cells with 3 or more autophagosomes were counted as having active autophagy and expressed as a percentage of total cells counted.
Vacuole preparation and bioanalyzer analysis
Vacuoles were purified from WT, rns2-2, and rns2-1 rosette leaves grown under short day conditions (Robert et al. 2007). Total Arabidopsis RNA was prepared using a Qiagen RNeasy plant mini kit (74904; Qiagen) and DNase treated using an on column DNaseI kit (79254; Qiagen). Purified vacuoles were flash-frozen in liquid nitrogen to induce membrane lysis. After thawing, 5 μg total RNA was incubated with the lysate for 20 min on ice and flash-frozen again in liquid nitrogen. RNA was extracted from the samples using a Qiagen RNeasy kit as above with a 30 μl elution volume. Samples were then diluted 50% and RNA analyzed using a 2100 Bioanalyzer (Agilent Technologies) and RNA nano chip (5067-1511; Agilent Technologies). Total RNA was quantified from chromatogram traces from 3 independent replicates using Bioanalyzer software with 200 nucleotides set as the minimum quantifiable size to avoid hydrolysis products produced by RNS2 or other RNases.
Results
rns2-1 encodes a truncated RNS2 protein
RNS2 contains an ER signal sequence at the N-terminus and is predicted to be secreted (Chou and Shen 2010), although its main localization is intracellular (Hillwig et al. 2011). As a number of plant vacuolar proteins contain vacuolar targeting signals at their C-terminus (Bednarek et al. 1990; Neuhaus et al. 1991), we hypothesized that the C-terminus of RNS2 may contain additional targeting information. The rns2-1 (SAIL_872_B10) and rns2-2 (SALK_069588) Arabidopsis T-DNA insertion mutants are both intron insertions (Fig. 1a). Whereas the rns2-2 insertion generates a null mutant (Hillwig et al. 2011; Floyd et al. 2015), the rns2-1 insertion is just upstream of the last exon and could potentially encode a truncated protein lacking the C-terminus. To assess this possibility, the 3′-end of the rns2-1 cDNA was amplified using 3′-RACE and an internal RNS2 primer upstream of the insertion site (Table 1). After sequencing (Supplementary Fig. S1), the 3′-region of rns2-1 was found to potentially encode a protein with two amino acid changes, D244G and G245K, followed by a premature stop codon (Fig. 1b). The C-terminal 14 amino acids were therefore eliminated, providing a tool to address the potential for a C-terminal vacuolar sorting signal, and the role of subcellular localization in RNS2 function.

An active RNase is produced in the rns2-1 mutant. a Schematic of rns2-1 and rns2-2 T-DNA insertion sites. Introns (lines), exons (boxes), start (arrow), and stop asterisk codons are indicated. b C-terminal predicted amino acid sequence of RNS2 and RNS2-1 proteins. Conserved sequence is highlighted in gray. Asterisk indicates a stop codon. c Activity gel of RNS2 activity. Total protein extracts from 7-day-old seedlings of the indicated genotypes were analyzed using an RNase activity in-gel assay. The position of RNS2 and RNS2-1 activity is indicated. d Confirmation of double mutant genotype. ATG9 expression was assessed by RT-PCR in WT, rns2-1, rns2-2, atg9-4, and rns2-1atg9-4 7-day-old seedlings. Arabidopsis 18S rRNA was used as a positive control
RNS2-1 is catalytically active
The rns2-2 null mutant lacks RNS2 RNase activity (Hillwig et al. 2011). To determine whether the rns2-1 mutant produces an active RNS2 RNase, RNS2 activity was analyzed by an in-gel RNase activity assay using high-molecular weight torula yeast RNA as substrate (Yen and Green 1991; Hillwig et al. 2011). rns2-1 was found to contain RNS2 RNase activity at a similar level to WT plants, while the rns2-2 negative control showed no RNS2 activity (Fig. 1c). However, the in-gel mobility of the RNS2-1 protein differed slightly from that of WT RNS2 (indicated by brackets in Fig. 1c), potentially as a result of altered post-translational modification. RNS2 is glycosylated (Hillwig et al. 2011), and different levels of glycosylation during synthesis results in multiple RNS2 bands on an activity gel (Taylor et al. 1993; Hillwig et al. 2011). It is possible that, although catalytically active, RNS2-1 may be mislocalized away from the machinery responsible for normal post-translational modification. Alternatively, the T-DNA insertion mutation could eliminate modification or processing sites, although putative glycosylation sites are not predicted in the C-terminal sequence.
rns2-1 has the same constitutive autophagy phenotype as rns2-2
Autophagy has been proposed to serve as a mechanism of rRNA and ribosome turnover in the cell (Kraft et al. 2008; Hillwig et al. 2011; Huang et al. 2015). The RNS2 null mutant, rns2-2, has increased basal autophagy (Hillwig et al. 2011), presumably as a mechanism to compensate for the lack of vacuolar RNA turnover (Floyd et al. 2015). To investigate whether plants containing the rns2-1 mutant allele have a similar phenotype, Arabidopsis seedlings grown under normal conditions were stained with MDC to visualize acidic vesicles, primarily autophagosomes, and roots were imaged by confocal microscopy (Fig. 2a). Autophagosomes were quantified by counting the number of MDC-stained structures per image, with the area in each image kept constant and expressed as autophagosomes per frame. Similar to rns2-2, rns2-1 showed a significant increase in MDC-stained structures compared to WT (Fig. 2b).

rns2-1 has constitutive autophagy that is dependent on ATG9. a Seven-day-old WT, rns2-1, rns2-2, atg9-4, and rns2-1atg9-4 Arabidopsis seedling roots were stained with 50 μM MDC and imaged using confocal microscopy. DIC indicates differential interference contrast microscopy. Scale bar 20 μm. b MDC-stained structures as in a were counted for each genotype, with at least 20 images counted for each genotype and three biological replicates. Autophagy activity is expressed as the mean number of autophagosomes (APs) detected per image (mean APs/frame). Asterisks indicate no significant difference according to the pairwise Student two-sided equal variance t test (P > 0.05). Error bars SE. c Protoplasts were prepared from WT, rns2-1 and rns2-2 plants and transformed with GFP-ATG8e as an autophagosome marker. Samples were treated with ConcA to prevent vacuolar degradation and the percentage of protoplasts with active autophagy, defined as the presence of three or more autophagic bodies, was determined. WT protoplasts were incubated with the known autophagy inducer tunicamycin as a positive control. Values are the mean of three independent replicates, with at least 25 protoplasts counted per replicate. Asterisks indicate no significant difference according to the pairwise Student two-sided equal variance t test (P > 0.05). Error bars SE. d rns2-1 mutants accumulate vesicles inside the vacuole. Seven-day-old WT, rns2-1, atg9-4, and rns2-1atg9-4 seedlings were treated for 8 h in liquid MS medium in the dark with DMSO as a solvent control or 1 μM ConcA to block vacuolar degradation. Roots were visualized using differential interference contrast microscopy. Insets show intravacuolar vesicle accumulation. Scale bar 20 μm for main figure and 6.5 μm for insets
To confirm that this increase represents an activation of autophagy, a GFP-ATG8e fusion protein was expressed in protoplasts from rns2-1 plants. ATG8e localizes to the membranes of autophagosomes in the cytoplasm and autophagic bodies in the vacuole (Contento et al. 2005) and therefore reflects the activity of the autophagy pathway. Protoplasts were treated with 1 µM ConcA to block degradation of autophagic bodies prior to quantification. ConcA inhibits V-type ATPases, raising the vacuolar pH. This inhibition subsequently reduces hydrolytic enzyme activities and blocks hydrolase delivery to the vacuole, resulting in intravacuolar accumulation of vesicles (Drose et al. 1993; Dettmer et al. 2006). The number of protoplasts with active autophagy, defined here as containing 3 or more autophagosomes or autophagic bodies (Liu et al. 2012), was substantially increased in rns2-1 compared with WT protoplasts, to a similar degree as in WT protoplasts treated with the autophagy inducer tunicamycin (as a positive control) or in the rns2-2 null mutant (Fig. 2c). Basal autophagy activity is therefore enhanced in the rns2-1 mutant, despite the presence of high levels of RNS2 activity.
To test the dependence of the rns2-1 increased autophagy phenotype on known autophagy genes, rns2-1 was crossed with the autophagy mutant atg9-4 [CS859902; (Floyd et al. 2015)] to generate an rns2-1atg9-4 double mutant. ATG9 (At2g31260) is a transmembrane protein involved in autophagy, probably in trafficking membrane lipids to newly forming autophagosomes, and is required for autophagosome formation (Noda et al. 2000; Hanaoka et al. 2002; Webber and Tooze 2010; Yamamoto et al. 2012). The rns2-1atg9-4 double mutant lines were confirmed to contain RNS2-1 RNase activity (Fig. 1c) and to lack ATG9 gene expression as expected (Fig. 1d). Unlike the rns2-1 single mutant, rns2-1atg9-4 lines had only very few MDC-stained puncta (less than 10 per image; Fig. 2a, b) and were statistically indistinguishable from WT (Fig. 2b), indicating that the observed fluorescent puncta in rns2-1 are autophagosomes generated by the classical autophagy pathway.
To confirm that the increased autophagosome phenotype seen in rns2-1 is the result of increased autophagosome formation rather than decreased autophagosome turnover, seedlings were treated with either 1 μM ConcA to block autophagic body degradation, or DMSO as a solvent control, for 8 h in liquid MS medium in darkness. As MDC staining depends on acidification of autophagosomes (Contento et al. 2005), and ConcA potentially inhibits acidification, MDC staining is not a reliable approach to visualize autophagosomes under these conditions. Vesicle accumulation in the vacuoles of WT, rns2-1, atg9-4, and rns2-1atg9-4 seedlings was therefore visualized by differential interference contrast microscopy, in which autophagic bodies, along with other vesicles, appear as raised puncta. In the presence of ConcA, rns2-1 seedlings accumulated substantially more intravacuolar vesicles than WT, with vesicles in WT likely resulting from basal autophagy and endocytosis (Fig. 2d). Vesicle accumulation was eliminated in atg9-4 and rns2-1atg9-4 lines, further supporting the hypothesis that the increased vesicle accumulation in rns2-1 results from autophagy. In the DMSO solvent controls, no vesicle accumulation was observed in any genotype (Fig. 2d), indicating that vesicle turnover is not affected in rns2-1 and that vesicle accumulation is therefore the result of increased formation of autophagosomes.
Complementation of rns2-1 mutant phenotype
To demonstrate that the mutant phenotype observed in rns2-1 is indeed due to the mutation in the RNS2 gene, the RNS2 cDNA under the control of the constitutive cauliflower mosaic virus 35S promoter (Floyd et al. 2015) was introduced into rns2-1 plants to assess whether it could complement the mutant phenotype. A FLAG tag was included in the RNS2 construct (Fig. 3a); however, we were unable to detect the protein using antibodies against the epitope tag, possibly due to cleavage of the tag from the RNS2 protein after synthesis. RNS2 activity in the resulting transgenic lines was assessed by in-gel RNase activity assay, and lines that expressed active RNS2 were selected (Fig. 3b). Transgenic seedlings were analyzed by MDC staining (Fig. 3c) to determine the extent to which expression of WT RNS2 could suppress the constitutive autophagy phenotype of the rns2-1 mutant. As expected, quantification by counting MDC-stained structures indicated that rns2-1 mutant seedlings had numerous MDC-stained puncta (approximately 20 per image), compared with few in control WT seedlings (approximately five per image; Fig. 3d). The WT RNS2 cDNA complemented this mutant phenotype, with the number of MDC-stained structures indistinguishable from WT plants (Fig. 3d). This indicates that the rns2-1 phenotype is due to loss of RNS2 cellular function, even though the enzymatic activity of the RNS2-1 protein is retained.

Complementation of rns2-1 mutant. a Construct design. A FLAG epitope tag was engineered into the RNS2 polypeptide after the signal peptide. Asterisk indicates position of stop codon. The RNS2-1 construct is a truncated version of RNS2 recreated using the sequence from Fig. 1. b FLAG-RNS2 was introduced into rns2-1 plants and FLAG-RNS2-1 into rns2-2 plants by Agrobacterium-mediated transformation. RNS2 activity in transgenic lines was assessed using an in-gel activity assay. c Seven-day-old seedlings of transgenic lines were stained with MDC, and roots were imaged using confocal microscopy. DIC indicates differential interference contrast microscopy. Scale bar 20 μm. d Autophagy activity in transgenic lines was estimated by counting MDC-stained structures visualized by fluorescence microscopy and compared with the parental genotypes. Means of three independent replicates are shown, with at least 20 images counted per replicate. Asterisks indicate no significant difference according to the pairwise Student two-sided equal variance t test (P > 0.05). Error bars = SE. e Protoplasts were prepared from WT, rns2-1, rns2-2, rns2-1 FLAGRNS2 and rns2-2 FLAGRNS2-1 lines and transformed with GFP-ATG8e as an autophagosome marker. Samples were treated with ConcA to prevent vacuolar degradation and observed under a fluorescence microscope. The percentage of protoplasts with active autophagy, defined as the presence of 3 or more autophagic bodies, was determined. Values are the means of 3 independent replicates, with at least 50 protoplasts counted per replicate. Asterisks indicate no significant difference according to the pairwise Student two-sided equal variance t test (P > 0.05). Error bars SE
To further confirm that the constitutive autophagy phenotype of rns2-1 was due to the loss of RNS2 function, the autophagosome marker GFP-ATG8e was expressed transiently in protoplasts from rns2-1 FLAGRNS2, rns2-2, rns2-1 and WT genotypes, followed by treatment with ConcA to block autophagosome degradation. As expected, the number of protoplasts with active autophagy in the rns2-1 and rns2-2 mutants was significantly higher than that of WT. Autophagy activity was restored to WT levels in rns2-1 FLAGRNS2 lines (Fig. 3e), confirming complementation of the mutant phenotype.
We have shown previously that the constitutive autophagy phenotype of the rns2-2 mutant can be complemented by the RNS2 WT cDNA (Floyd et al. 2015). We hypothesized that, in contrast to the WT cDNA, the RNS2-1 mutant cDNA would be unable to complement the rns2-2 null mutant due to its inability to function appropriately in a cellular context. To test this hypothesis, a cDNA encoding the predicted RNS2-1 truncated protein (Fig. 3a) was expressed from a 35S promoter in rns2-2 null mutant plants and its RNase activity verified (Fig. 3b). The 35S::RNS2-1 construct was unable to complement the rns2-2 mutant phenotype, determined either by MDC staining (Fig. 3c, d) or GFP-ATG8e expression (Fig. 3e), indicating that the truncated protein produced by the rns2-1 mutant cannot fulfill the requisite RNS2 function.
RNS2-1 shows reduced localization to the vacuole
The rns2-1 phenotypes observed are equivalent to those seen in the rns2-2 null mutant. Since rns2-1 is not a null mutant and retains high RNS2 catalytic activity, we hypothesized that the RNS2-1 protein is mislocalized, thus resulting in the mutant phenotype. RNS2 activity is found in both ER and vacuole fractions, but is not an acidic RNase, showing its highest catalytic activity near pH 7.5 (Hillwig et al. 2011). It is therefore unclear whether RNS2′s catalytic function as an RNase is required in the acidic vacuole. In the absence of RNS2, rRNA accumulates within the vacuole (Floyd et al. 2015), indicating a potential function for RNA degradation within this organelle. While C-terminal vacuolar sorting signals can direct proteins to the vacuole (Bednarek et al. 1990; Neuhaus et al. 1991; Saalbach et al. 1996), they share little homology in sequence or size, making their computational identification difficult. Loss of vacuolar localization of RNS2 in the rns2-1 mutant may explain the observed increased basal autophagy phenotype.
To address this possibility, RNase activity within vacuoles from rns2 mutants and WT plants was assessed. WT, rns2-1, and rns2-2 vacuoles were purified from rosette leaves and the integrity of the isolated vacuoles was determined by measuring the activity of acid phosphatase, a vacuolar marker enzyme, as in Floyd et al. (2015). Vacuoles from all genotypes had similar activity (Supplementary Fig. S2a), and equal amounts of vacuoles were used in all later experiments. Purity of the vacuole preparations was determined by immunoblotting using antibodies against the vacuolar protein aleurain (Ahmed et al. 2000) and the trans-Golgi network protein SYP41 (Sanderfoot et al. 2001). Aleurain co-purified with the vacuoles, whereas SYP41 was present in total protoplast protein samples but absent from the vacuole preparations (Supplementary Fig. S2b).
RNS2 activity in total protoplast and isolated vacuole samples from each genotype was determined using an in-gel RNase activity assay (Supplementary Fig. S2c). Activity at the migration position expected for RNS2 was detected in protoplasts and vacuoles for WT plants, whereas, as expected, little activity was seen in the null mutant rns2-2. Activity was present in the protoplast sample from rns2-1 (note that the lane is distorted due to the high protein concentration), but the activity in the rns2-1 vacuoles was almost undetectable, indicating that the majority of the activity is present elsewhere, potentially in the ER or other endomembrane organelles.
To demonstrate quantitatively that rns2-1 vacuoles lacked high RNS2 activity, vacuoles were lysed by freezing and incubated with 5 μg of Arabidopsis total RNA. RNA degradation was quantified using a Bioanalyzer. As RNS2 is the only RNase identified within vacuoles (Carter et al. 2004), any RNA degradation in the vacuole sample is expected to be due mainly to RNS2. WT vacuoles were used as a positive control since they contain RNS2 RNase and therefore are expected to degrade the added RNA. rns2-2 vacuoles were used as a negative control since RNS2 activity is missing, and therefore any RNA degradation seen must be due either to other unidentified RNases present in the vacuole, or to contamination from other, non-vacuolar, RNases. As RNS2-1 protein is fully catalytically active, if present in the vacuole then RNA degradation should occur to a similar extent as in WT samples. Alternatively, if RNS2-1 is not vacuole-localized, then RNA degradation would resemble that of rns2-2. Peak areas from output traces from the Bioanalyzer were quantified (Fig. 4a, c) and also represented as computationally-generated gel images for ease of visualization (Fig. 4b). WT vacuole extracts completely degraded the added RNA. By contrast, rns2-1 vacuole extracts showed reduced RNA degradation, statistically indistinguishable from that of rns2-2 mutants, indicating that RNS2-1 activity is not localized within the vacuole.

RNS2-1 activity does not localize to the vacuole. a Vacuoles were isolated from the indicated genotypes, lysed, and the lysates incubated with total Arabidopsis RNA. Remaining un-degraded RNA was quantified using Bioanalyzer traces with a minimum setting of 200 nucleotides. RNA alone (no vacuole extract) was used as a positive control and results are expressed relative to the RNA alone average. Each sample was analyzed in triplicate for each of three biological replicates. Error bars represent SE. Similar letters indicate no significant difference according to pairwise Student’s two-sided t test (P > 0.05). b Representative Bioanalyzer chromatogram. Genotypes are indicated at top. c Representative Bioanalyzer output traces for each genotype as used for quantification. The area under each peak was determined for three biological replicates and represented graphically in a
These findings also suggest that a vacuolar localization signal exists in the last 16 amino acids at the RNS2 C-terminus, and that loss of this signal in rns2-1 disrupts its vacuolar localization. Furthermore, the loss of RNS2 activity in the vacuole of rns2-1 plants is likely to result in the increased autophagy phenotype as seen for the rns2-2 null mutant (Hillwig et al. 2011).
RNS2-1 is exported from the cell
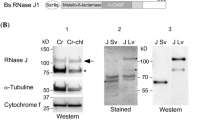
RNS2 contains an N-terminal signal peptide that targets it into the ER (Taylor et al. 1993; Hillwig et al. 2011). Proteins targeted to the secretory pathway are secreted from the cell unless they harbor an organelle retention or localization signal (Vitale and Denecke 1999). We have shown that the C-terminal mutation present in rns2-1 disrupts vacuole localization, and we hypothesize that the protein would therefore be secreted from the cell. To test this, a FLAG epitope tag was inserted between the N-terminal RNS2 signal peptide and the mature portion of RNS2 WT and RNS2-1 mutant proteins (Einhauer and Jungbauer 2001) (Fig. 3a). These constructs were transiently expressed in WT leaf protoplasts. After 44 h incubation, samples were divided into cell and medium fractions and immunoblotted using FLAG antibodies. RNS2 remained in the cellular fraction as expected, while a portion of RNS2-1 was exported out of the cell (Fig. 5). The vacuolar protease aleurain was found in cell fractions in both samples as expected (Fig. 5). This indicates that disruption of the C-terminus of RNS2 results in secretion of the protein after transit through the ER rather than targeting to the vacuole, indicating that RNS2 activity in the vacuole is required for its physiological function.

FLAG-RNS2-1 is exported from the cell. Protoplasts from WT plants were transiently transformed with FLAG-RNS2 or FLAG-RNS2-1 and incubated for 44 h to allow expression. Proteins were fractionated into cell (Cell) and medium (Med) fractions and analyzed by immunoblotting using FLAG antibodies, and aleurain antibodies as a control. Image shows a representative of four biological replicates
The C-terminal extension is conserved in class II RNase T2 enzymes
Analysis of the C-terminus of plant Class I and Class II RNase T2 proteins showed that Class II enzymes have an extension that is not found in Class I (Fig. 6). The C-terminal tail varies in length, from 13 to 27 aa, among the sequences analyzed. These tails do not have conserved motifs; however, all contain a large proportion of charged or polar amino acids. This Class II tail is conserved in gymnosperms and angiosperms, including dicots and monocots. Class II proteins with known subcellular localization are found in the ER or the vacuole and possess the C-terminal extension, while Class I enzymes with confirmed localization are secreted to the apoplast and do not possess this tail. The conservation of the C-terminal extension is consistent with a functional role for this feature in Class II proteins, and supports the hypothesis that vacuolar localization signals necessary for Class II biological function are present in this region of the protein.

The C-terminal extension is conserved in class II RNase T2 enzymes. C-terminal sequences for representative Class I and Class II RNase T2 plant proteins were aligned using the most C-terminal conserved cysteine residue as reference. Polar (green) or charged (red negative; blue positive) amino acid residues in the C-terminal extension are highlighted. Experimentally determined subcellular localization (ER endoplasmic reticulum, VAC vacuole, AP apoplast, AP* extracellular digestive secretion) is indicated on the right for RNase LX (Lehmann et al. 2001), RNase LE (Jost et al. 1991), RNase NE (Hugot et al. 2002), RNS1 and RNS2 (Bariola et al. 1999; Hillwig et al. 2011), RNase DA-I (Okabe et al. 2005), and RNase LER (Kothke and Kock 2011). Protein nomenclature follows MacIntosh et al. (2010)
An interesting exception to this general difference between the two classes of plant RNase T2 proteins is a small subset of Class I enzymes with a short C-terminal extension not found in other Class I proteins (Fig. 6). These proteins from tomato, tobacco, and zinnia have a short (9 aa) extension that also contains charged and polar amino acids. Unlike most Class I enzymes, these proteins are predicted or have been found to localize to the ER and vacuole.
Finally, while Class II proteins have been proposed as the ancestral form of RNase T2 in plants (MacIntosh et al. 2010), a Class II protein seems to be missing in the genome of Physcomitrella patens. The genome of this moss contains two RNase T2 genes, PpRNS1 and PpRNS3, but both group with the Class I clade (MacIntosh et al. 2010), suggesting that the ancestral Class II gene was lost during evolution of this species. However, closer analysis of the predicted protein sequence for the two moss genes showed that PpRNS3 has a 26 aa C-terminal extension rich in polar and charged residues, similar to the tails of Class II proteins (Fig. 6). This finding suggests that a Class I protein acquired Class II properties in this moss, and explains how the Class II RNase T2 gene that seems essential to maintain cellular homeostasis was lost in this species.
Discussion
RNase T2 enzymes represent a family of RNases that either are secreted or are localized intracellularly within the vacuole or lysosome (Irie 1999). Similar 2′-3′-cyclizing RNase groups, RNase T1 and RNase A, exist in fungal, bacterial, and animal species, but are absent from plants (Yoshida 2001; Dyer and Rosenberg 2006). However, the RNase T2 family represents the most widely distributed of these RNases and is conserved in the majority of genomes analyzed (MacIntosh 2011). While the Arabidopsis genome contains five RNase T2 genes (Taylor and Green 1991; Igic and Kohn 2001), RNS2 is the only family member expressed in all tissues and developmental stages tested and this expression pattern is conserved for other members of the plant Class II RNase T2 family, suggesting it may serve an important housekeeping role (Taylor et al. 1993; MacIntosh et al. 2010). RNS2 is found within the ER and vacuole, locations that are not typically associated with the presence of RNA, raising the question of its substrates and physiological function. The null mutant rns2-2 has elevated basal autophagy and we hypothesized that this phenotype is a compensatory mechanism to cope with the lack of normal rRNA degradation by RNS2 in the vacuole (Hillwig et al. 2011; Floyd et al. 2015). However, the possibility that the loss of a non-vacuolar function of RNS2 in the mutant is the cause of the phenotype could not be discarded in previous experiments.
Here, we have shown that localization of RNS2 to the vacuole is necessary to maintain cellular homeostasis in Arabidopsis. Secretion of RNS2 to the apoplast after ER targeting in the rns2-1 mutant, rather than traffic to the vacuole, results in a constitutive autophagy phenotype, despite full catalytic activity of the protein. This phenotype is similar to that observed in rns2-2 null mutants, suggesting that mislocalization of the RNS2-1 protein prevents its function in rRNA recycling. We have previously shown that rns2-2 mutants accumulate rRNA inside the vacuole and that this accumulation is lost in some mutants defective in autophagy (Floyd et al. 2015). Loss of vacuolar RNS2 activity therefore results in upregulation of autophagy as a compensatory mechanism of RNA decay, and secreted RNS2 cannot substitute for vacuolar activity. Despite these substantial molecular and cellular phenotypes, both the rns2-1 and rns2-2 mutants are developmentally and morphologically very similar to WT plants, indicating that the mutants are able to compensate for the loss of the RNase activity. Based on these data, together with those of Hillwig et al. (2011) and Floyd et al. (2015), we hypothesize that rRNA is transported into the vacuole by an autophagy-like mechanism, where it is degraded by RNS2.
Our analysis of the C-terminally truncated RNS2-1 protein suggests that the C-terminus of the protein constitutes a vacuolar targeting signal. C-terminal propeptides have been shown to function in vacuolar sorting in a range of proteins, and these signals are poorly conserved in sequence and thus difficult to predict computationally (Bednarek et al. 1990; Neuhaus et al. 1991; Saalbach et al. 1996; Koide et al. 1999; Cervelli et al. 2004; Tapernoux-Luthi et al. 2007). Proteins containing these C-terminal signals are recognized by vacuolar sorting receptors for trafficking to the vacuole. The recognition event probably occurs at the trans-Golgi network, followed by receptor-mediated transport to the prevacuolar compartment and then on to the vacuole (Sanderfoot et al. 1998; Kang et al. 2012; Fuji et al. 2016). There has been some debate in the literature on the itinerary of these receptors, with suggestions that they may cycle between ER and Golgi rather than functioning at the trans-Golgi network (Niemes et al. 2010a, b). In any case, we predict that the C-terminus of RNS2 is recognized by a member(s) of the vacuolar sorting receptor family and selected for transport to the vacuole. Based on comparisons to other Class II sequences, it seems evident that this recognition is not based on a specific amino acid sequence. Rather, a tail of variable size, rich in polar and charged amino acids, may serve as the signal recognized by the vacuolar transport machinery.
Upon overexpression as a CFP fusion protein, RNS2 is seen in ER bodies in addition to the vacuole (Hillwig et al. 2011). ER bodies are rod-shaped structures that form in Arabidopsis and other Brassicales species and are contiguous with the ER network (Nakano et al. 2014). They are abundant in seedlings and can be induced in older leaves by wounding and other stresses, suggesting that they may be involved in defense (Matsushima et al. 2002). The observation that RNS2 has the highest activity at neutral pH (Hillwig et al. 2011) suggests that ER body-localized RNS2 could also be active in addition to vacuole-localized enzyme; however, the potential substrate for a RNase inside the ER is unclear, and there is no evidence that RNS2 has a role in defense responses. Alternatively, it is possible that the ER body localization is an artifact of overexpression; although endogenous RNS2 activity is also found in cellular fractions containing ER (Hillwig et al. 2011), it is not known whether it is in ER bodies. As RNS2 must traffic through the ER to reach the vacuole, endogenous activity within the ER could reflect newly synthesized protein in transit to the vacuole. While not at its maximal activity at the pH of the vacuole, RNS2 has a broad pH optimum and is expected to still exhibit substantial activity at acidic pH (Hillwig et al. 2011), which is apparently sufficient for its physiological function within this organelle.
Sequence comparisons indicate that the C-terminal tail containing putative vacuolar localization signals is conserved in Class II proteins, and it is consistent with the conserved housekeeping role in RNA metabolism and cellular homeostasis assigned to this class of RNase T2 enzymes. The identification of a Class I protein that has acquired a Class II-like tail in the moss Physcomitrella patens, a species that lacks a gene encoding a Class II enzyme (MacIntosh et al. 2010), also supports the idea that an RNase T2 protein functioning in the vacuole is necessary to maintain cellular homeostasis.
Additionally, a few Class I proteins in angiosperms seem to have acquired a different C-terminal extension that also targets proteins to the ER or the vacuole. RNase LX from tomato is not normally expressed, but it is strongly induced in response to phosphate starvation, and during developmental processes that involve programmed cell death such as tracheary element differentiation and senescence (Lehmann et al. 2001). The C-terminal extension in this protein contains a HDEF motif that serves as ER-retention signal, and the protein has been found localized to the ER, while two posttranslational processing products of the protein have been found in the vacuole (Loffler et al. 1992; Lehmann et al. 2001). Subcellular localization for the other Class I RNases with similar tails has not been determined. However, tobacco NGR3 is only expressed in leaves in response to viral infections (Kurata et al. 2002), while ZnRNaseI is expressed in zinnia leaves only during tracheary element differentiation (Ye and Droste 1996). It is important to note that although RNS2 is constitutively expressed at high levels in most plant tissues and organs, its expression is further induced by phosphate starvation and during senescence (Taylor et al. 1993; Bariola et al. 1999; MacIntosh et al. 2010). Whether this regulation indicates that RNS2 carries an additional function common also to these Class I enzymes and to other phosphate-regulated and ER-localized RNases (Rojas et al. 2015), and perhaps related to its ER localization, is not known.
In summary, our data indicate that the RNase RNS2 is transported to the vacuole via a pathway dependent on a C-terminal targeting signal. Disruption of this signal results in secretion of the enzyme, causing a phenotype similar to that of a null mutant, despite retention of full enzymatic activity. The phenotype includes the constitutive activation of autophagy, presumably in an attempt to compensate for loss of vacuolar RNA degradation. Future work will assess the mechanism by which autophagy is activated in the mutant and the physiological consequences of this activation.
Author contributions statement
DCB and GCM conceived and designed research. BEF, YM and SCM conducted experiments. BEF, DCB and GCM analyzed data. BEF and DCB wrote the manuscript. All authors revised the manuscript and read and approved the final version.
Abbreviations
- ConcA:
-
Concanamycin A
- ER:
-
Endoplasmic reticulum
- MDC:
-
Monodansylcadaverine
- WT:
-
Wild-type
References
Ahmed SU, Rojo E, Kovaleva V, Venkataraman S, Dombrowski JE, Matsuoka K, Raikhel NV (2000) The plant vacuolar sorting receptor AtELP is involved in transport of NH2-terminal propeptide-containing vacuolar proteins in Arabidopsis thaliana. J Cell Biol 149:1335–1344. doi:10.1083/jcb.149.7.1335
Andersen KL, Collins K (2012) Several RNase T2 enzymes function in induced tRNA and rRNA turnover in the ciliate Tetrahymena. Mol Biol Cell 23(1):36–44. doi:10.1091/mbc.E11-08-0689
Andersen KR, Jensen TH, Brodersen DE (2008) Take the “A” tail–quality control of ribosomal and transfer RNA. Biochim Biophys Acta 1779(9):532–537. doi:10.1016/j.bbagrm.2008.06.011
Balagopal V, Parker R (2009) Polysomes, P bodies and stress granules: states and fates of eukaryotic mRNAs. Curr Opin Cell Biol 21(3):403–408. doi:10.1016/j.ceb.2009.03.005
Bariola PA, Howard CJ, Taylor CB, Verburg MT, Jaglan VD, Green PJ (1994) The Arabidopsis ribonuclease gene RNS1 is tightly controlled in response to phosphate limitation. Plant J 6(5):673–685
Bariola PA, MacIntosh GC, Green PJ (1999) Regulation of S-like ribonuclease levels in Arabidopsis. Antisense inhibition of RNS1 or RNS2 elevates anthocyanin accumulation. Plant Physiol 119(1):331–342
Bednarek SY, Wilkins TA, Dombrowski JE, Raikhel NV (1990) A carboxyl-terminal propeptide is necessary for proper sorting of barley lectin to vacuoles of tobacco. Plant Cell 2(12):1145–1155. doi:10.1105/tpc.2.12.1145
Carter C, Pan S, Zouhar J, Avila E, Girke T, Raikhel N (2004) The vegetative vacuole proteome of Arabidopsis thaliana reveals predicted and unexpected proteins. Plant Cell 16(12):3285–3303
Cervelli M, Di Caro O, Di Penta A, Angelini R, Federico R, Vitale A, Mariottini P (2004) A novel C-terminal sequence from barley polyamine oxidase is a vacuolar sorting signal. Plant J 40(3):410–418. doi:10.1111/j.1365-313X.2004.02221.x
Chou KC, Shen HB (2010) Plant-mPLoc: a top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS One 5(6):e11335. doi:10.1371/journal.pone.0011335
Clough S, Bent A (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16(6):735–743
Condon C, Putzer H (2002) The phylogenetic distribution of bacterial ribonucleases. Nucleic Acids Res 30(24):5339–5346
Contento AL, Xiong Y, Bassham DC (2005) Visualization of autophagy in Arabidopsis using the fluorescent dye monodansylcadaverine and a GFP-AtATG8e fusion protein. Plant J 42(4):598–608
Dettmer J, Hong-Hermesdorf A, Stierhof YD, Schumacher K (2006) Vacuolar H+ -ATPase activity is required for endocytic and secretory trafficking in Arabidopsis. Plant Cell 18(3):715–730. doi:10.1105/tpc.105.037978
Drose S, Bindseil KU, Bowman EJ, Siebers A, Zeeck A, Altendorf K (1993) Inhibitory effect of modified bafilomycins and concanamycins on P- and V-type adenosinetriphosphatases. Biochemistry 32(15):3902–3906
Dyer KD, Rosenberg HF (2006) The RNase A superfamily: generation of diversity and innate host defense. Mol Divers 10(4):585–597. doi:10.1007/s11030-006-9028-2
Einhauer A, Jungbauer A (2001) The FLAG peptide, a versatile fusion tag for the purification of recombinant proteins. J Biochem Biophys Methods 49(1–3):455–465
Floyd BE, Morriss SC, Macintosh GC, Bassham DC (2012) What to eat: evidence for selective autophagy in plants. J Integr Plant Biol 54(11):907–920. doi:10.1111/j.1744-7909.2012.01178.x
Floyd BE, Morriss SC, MacIntosh GC, Bassham DC (2015) Evidence for autophagy-dependent pathways of rRNA turnover in Arabidopsis. Autophagy 11(12):2199–2212. doi:10.1080/15548627.2015.1106664
Fuji K, Shirakawa M, Shimono Y, Kunieda T, Fukao Y, Koumoto Y, Takahashi H, Hara-Nishimura I, Shimada T (2016) The adaptor complex AP-4 regulates vacuolar protein sorting at the trans-Golgi network by interacting with VACUOLAR SORTING RECEPTOR1. Plant Physiol 170(1):211–219. doi:10.1104/pp.15.00869
Hanaoka H, Noda T, Shirano Y, Kato T, Hayashi H, Shibata D, Tabata S, Ohsumi Y (2002) Leaf senescence and starvation-induced chlorosis are accelerated by the disruption of an Arabidopsis autophagy gene. Plant Physiol 129(3):1181–1193
Haud N, Kara F, Diekmann S, Henneke M, Willer JR, Hillwig MS, Gregg RG, Macintosh GC, Gartner J, Alia A, Hurlstone AF (2011) rnaset2 mutant zebrafish model familial cystic leukoencephalopathy and reveal a role for RNase T2 in degrading ribosomal RNA. Proc Natl Acad Sci USA 108(3):1099–1103. doi:10.1073/pnas.1009811107
Henneke M, Diekmann S, Ohlenbusch A, Kaiser J, Engelbrecht V, Kohlschutter A, Kratzner R, Madruga-Garrido M, Mayer M, Opitz L, Rodriguez D, Ruschendorf F, Schumacher J, Thiele H, Thoms S, Steinfeld R, Nurnberg P, Gartner J (2009) RNASET2-deficient cystic leukoencephalopathy resembles congenital cytomegalovirus brain infection. Nat Genet 41(7):773–775. doi:10.1038/ng.398
Hillwig MS, Rizhsky L, Wang Y, Umanskaya A, Essner JJ, MacIntosh GC (2009) Zebrafish RNase T2 genes and the evolution of secretory ribonucleases in animals. BMC Evol Biol 9:170. doi:10.1186/1471-2148-9-170
Hillwig MS, Contento AL, Meyer A, Ebany D, Bassham DC, Macintosh GC (2011) RNS2, a conserved member of the RNase T2 family, is necessary for ribosomal RNA decay in plants. Proc Natl Acad Sci USA 108(3):1093–1098. doi:10.1073/pnas.1009809108
Houseley J, Tollervey D (2009) The many pathways of RNA degradation. Cell 136(4):763–776. doi:10.1016/j.cell.2009.01.019
Hua ZH, Fields A, Kao TH (2008) Biochemical models for S-RNase-based self-incompatibility. Mol Plant 1(4):575–585. doi:10.1093/mp/ssn032
Huang H, Kawamata T, Horie T, Tsugawa H, Nakayama Y, Ohsumi Y, Fukusaki E (2015) Bulk RNA degradation by nitrogen starvation-induced autophagy in yeast. EMBO J 34(2):154–168. doi:10.15252/embj.201489083
Hugot K, Ponchet M, Marais A, Ricci P, Galiana E (2002) A tobacco S-like RNase inhibits hyphal elongation of plant pathogens. Mol Plant Microbe Interact 15(3):243–250. doi:10.1094/mpmi.2002.15.3.243
Igic B, Kohn JR (2001) Evolutionary relationships among self-incompatibility RNases. Proc Natl Acad Sci USA 98(23):13167–13171. doi:10.1073/pnas.231386798
Irie M (1999) Structure-function relationships of acid ribonucleases: lysosomal, vacuolar, and periplasmic enzymes. Pharmacol Ther 81(2):77–89
Jost W, Bak H, Glund K, Terpstra P, Beintema JJ (1991) Amino acid sequence of an extracellular, phosphate-starvation-induced ribonuclease from cultured tomato (Lycopersicon esculentum) cells. Eur J Biochem 198(1):1–6
Kang H, Kim SY, Song K, Sohn EJ, Lee Y, Lee DW, Hara-Nishimura I, Hwang I (2012) Trafficking of vacuolar proteins: the crucial role of Arabidopsis vacuolar protein sorting 29 in recycling vacuolar sorting receptor. Plant Cell 24(12):5058–5073. doi:10.1105/tpc.112.103481
Koide Y, Matsuoka K, Ohto M, Nakamura K (1999) The N-terminal propeptide and the C terminus of the precursor to 20-kilo-dalton potato tuber protein can function as different types of vacuolar sorting signals. Plant Cell Physiol 40(11):1152–1159
Kothke S, Kock M (2011) The Solanum lycopersicum RNaseLER is a class II enzyme of the RNase T2 family and shows preferential expression in guard cells. J Plant Physiol 168(8):840–847. doi:10.1016/j.jplph.2010.11.012
Kraft C, Deplazes A, Sohrmann M, Peter M (2008) Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol 10(5):602–610. doi:10.1038/ncb1723
Kurata N, Kariu T, Kawano S, Kimura M (2002) Molecular cloning of cDNAs encoding ribonuclease-related proteins in Nicotiana glutinosa leaves, as induced in response to wounding or to TMV-infection. Biosci Biotechnol Biochem 66(2):391–397. doi:10.1271/bbb.66.391
Lehmann K, Hause B, Altmann D, Kock M (2001) Tomato ribonuclease LX with the functional endoplasmic reticulum retention motif HDEF is expressed during programmed cell death processes, including xylem differentiation, germination, and senescence. Plant Physiol 127(2):436–449
Liu Y, Burgos JS, Deng Y, Srivastava R, Howell SH, Bassham DC (2012) Degradation of the endoplasmic reticulum by autophagy during endoplasmic reticulum stress in Arabidopsis. Plant Cell 24(11):4635–4651. doi:10.1105/tpc.112.101535
Loffler A, Abel S, Jost W, Beintema JJ, Glund K (1992) Phosphate-regulated induction of intracellular ribonucleases in cultured tomato (Lycopersicon esculentum) cells. Plant Physiol 98(4):1472–1478
MacIntosh GC (2011) RNase T2 Family: Enzymatic properties, functional diversity, and evolution of ancient ribonucleases. In: Nicholson AWW (ed) ribonucleases, vol 26. Springer, Berlin Heidelberg, pp 89–114
MacIntosh GC, Bariola PA, Newbigin E, Green PJ (2001) Characterization of Rny1, the Saccharomyces cerevisiae member of the T2 RNase family of RNases: unexpected functions for ancient enzymes? Proc Natl Acad Sci USA 98(3):1018–1023. doi:10.1073/pnas.98.3.1018
MacIntosh GC, Hillwig MS, Meyer A, Flagel L (2010) RNase T2 genes from rice and the evolution of secretory ribonucleases in plants. Mol Genet Genomics 283(4):381–396. doi:10.1007/s00438-010-0524-9
Matsushima R, Hayashi Y, Kondo M, Shimada T, Nishimura M, Hara-Nishimura I (2002) An endoplasmic reticulum-derived structure that is induced under stress conditions in Arabidopsis. Plant Physiol 130(4):1807–1814. doi:10.1104/pp.009464
Meng X, Sun P, Kao TH (2011) S-RNase-based self-incompatibility in Petunia inflata. Ann Bot 108(4):637–646. doi:10.1093/aob/mcq253
Nakano RT, Yamada K, Bednarek P, Nishimura M, Hara-Nishimura I (2014) ER bodies in plants of the Brassicales order: biogenesis and association with innate immunity. Front Plant Sci 5:73. doi:10.3389/fpls.2014.00073
Neuhaus JM, Sticher L, Meins F Jr, Boller T (1991) A short C-terminal sequence is necessary and sufficient for the targeting of chitinases to the plant vacuole. Proc Natl Acad Sci USA 88(22):10362–10366
Niemes S, Labs M, Scheuring D, Krueger F, Langhans M, Jesenofsky B, Robinson DG, Pimpl P (2010a) Sorting of plant vacuolar proteins is initiated in the ER. Plant J 62(4):601–614
Niemes S, Langhans M, Viotti C, Scheuring D, San Wan Yan M, Jiang L, Hillmer S, Robinson DG, Pimpl P (2010b) Retromer recycles vacuolar sorting receptors from the trans-Golgi network. Plant J 61(1):107–121. doi:10.1111/j.1365-313X.2009.04034.x
Noda T, Kim J, Huang WP, Baba M, Tokunaga C, Ohsumi Y, Klionsky DJ (2000) Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J Cell Biol 148(3):465–480
Okabe T, Yoshimoto I, Hitoshi M, Ogawa T, Ohyama T (2005) An S-like ribonuclease gene is used to generate a trap-leaf enzyme in the carnivorous plant Drosera adelae. FEBS Lett 579(25):5729–5733. doi:10.1016/j.febslet.2005.09.043
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8(10):785–786. doi:10.1038/nmeth.1701
Robert S, Zouhar J, Carter C, Raikhel N (2007) Isolation of intact vacuoles from Arabidopsis rosette leaf-derived protoplasts. Nat Protoc 2(2):259–262
Rojas H, Floyd B, Morriss S, Bassham D, MacIntosh G, Goldraij A (2015) NnSR1, a class III non-S-RNase specifically induced in Nicotiana alata under Pi deficiency, is localized in endoplasmic reticulum compartments. Plant Sci 236:250–259
Saalbach G, Rosso M, Schumann U (1996) The vacuolar targeting signal of the 2S albumin from Brazil nut resides at the C terminus and involves the C-terminal propeptide as an essential element. Plant Physiol 112(3):975–985
Sanderfoot A, Ahmed S, Marty-Mazars D, Rapoport I, Kirchhausen T, Marty F, Raikhel N (1998) A putative vacuolar cargo receptor partially colocalizes with AtPEP12p on a prevacuolar compartment in Arabidopsis roots. Proc Natl Acad Sci USA 95(17):9920–9925
Sanderfoot AA, Kovaleva V, Bassham DC, Raikhel NV (2001) Interactions between syntaxins identify at least five SNARE complexes within the Golgi/prevacuolar system of the Arabidopsis cell. Mol Biol Cell 12(12):3733–3743
Sheen J (2002) A transient expression assay using Arabidopsis mesophyll protoplasts. Available online at http://molbio.mgh.harvard.edu/sheenweb/protocols_reg.html
Tapernoux-Luthi EM, Schneider T, Keller F (2007) The C-terminal sequence from common bugle leaf galactan:galactan galactosyltransferase is a non-sequence-specific vacuolar sorting determinant. FEBS Lett 581(9):1811–1818. doi:10.1016/j.febslet.2007.03.068
Taylor CB, Green PJ (1991) Genes with homology to fungal and S-Gene RNases are expressed in Arabidopsis thaliana. Plant Physiol 96(3):980–984
Taylor CB, Bariola PA, delCardayre SB, Raines RT, Green PJ (1993) RNS2: a senescence-associated RNase of Arabidopsis that diverged from the S-RNases before speciation. Proc Natl Acad Sci USA 90(11):5118–5122
Vitale A, Denecke J (1999) The endoplasmic reticulum-gateway of the secretory pathway. Plant Cell 11(4):615–628
Webber JL, Tooze SA (2010) New insights into the function of Atg9. FEBS Lett 584(7):1319–1326. doi:10.1016/j.febslet.2010.01.020
Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, Ichikawa R, Kinjo M, Ohsumi Y (2012) Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol 198(2):219–233. doi:10.1083/jcb.201202061
Yang X, Bassham DC (2015) New insight into the mechanism and function of autophagy in plant cells. Int Rev Cell Mol Biol 320:1–40. doi:10.1016/bs.ircmb.2015.07.005
Ye ZH, Droste DL (1996) Isolation and characterization of cDNAs encoding xylogenesis-associated and wounding-induced ribonucleases in Zinnia elegans. Plant Mol Biol 30(4):697–709
Yen Y, Green PJ (1991) Identification and properties of the major ribonucleases of Arabidopsis thaliana. Plant Physiol 97(4):1487–1493
Yoshida H (2001) The ribonuclease T1 family. Methods Enzymol 341:28–41
Acknowledgements
This work was supported by Grant No. MCB-1051818 from the United States National Science Foundation to GCM and DCB and Grant No. DE-SC0014038 from the United States Department of Energy to DCB. We thank Danielle Ebany for isolation of an rns2-1 homozygote and Junmarie Soto-Burgos for assistance with confocal microscopy.
Author information
Authors and Affiliations
Corresponding authors
Additional information
B. E. Floyd and Y. Mugume contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Floyd, B.E., Mugume, Y., Morriss, S.C. et al. Localization of RNS2 ribonuclease to the vacuole is required for its role in cellular homeostasis. Planta 245, 779–792 (2017). https://doi.org/10.1007/s00425-016-2644-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-016-2644-x