
Abstract
Main conclusion
We used transcript profiling and multi-SNP association to investigate the genetic regulatory relationship between miRNA Pto-miR530a and its target Pto-KNAT1, identifying additive, dominant, and epistatic effects.
MicroRNAs (miRNAs) play crucial roles in the post-transcriptional regulation of plant growth and development; indeed, many studies have described the importance of miRNA–target interactions in herbaceous species. However, elucidation of the miRNA–target interactions in trees may require novel strategies. In the present study, we describe a strategy combining expression profiling by reverse transcription quantitative PCR (RT-qPCR) and association mapping with multiple single nucleotide polymorphisms (SNPs) to evaluate the interaction between Pto-miR530a and its target Pto-KNAT1 in Populus tomentosa. RT-qPCR analysis showed a negative correlation (r = −0.62, P < 0.05) between expression levels of Pto-miR530a and Pto-KNAT1 in eight tissues. We used a Bayesian hierarchical model to identify allelic variants of Pto-miR530a and Pto-KNAT1 that associated with eight traits related to growth and wood properties, in a population of 460 unrelated individuals of P. tomentosa. This analysis identified 27 associations, with the proportions of phenotypic variance (R 2) contributed by each SNP ranging of 0.82–15.81 %, the additive effects of each SNP ranging of 0.16–18.09, and the dominant effects ranging from −14.09 to 19.00. Epistatic interaction models showed a strong interaction among SNPs in the miRNA target with R 2 of 0.1–3.56 %, and information gain of significant SNP pairs of −3.09 to 0.93 %, representing the regulatory interactions between the miRNA and the mRNA. Thus, we used a new strategy that combines association genetics and expression profiling based on SNPs to study the regulatory relationship between this miRNA and its target mRNA, thereby providing novel advances in our understanding of the genetic architecture of important traits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs) are endogenous, noncoding, small RNAs (20–24 nt) that regulate gene transcript levels by cleavage or post-transcriptional inhibition of their target mRNAs (Chen 2004; Jones-Rhoades et al. 2006). Therefore, the biological function of each miRNA depends mainly on its target genes. More than 35,000 miRNAs have been identified from various species and computational algorithms have predicted thousands of targets to date, but only a handful of targets have been experimentally validated (Kozomara and Griffiths-Jones 2014). Previous studies revealed the biological importance of miRNA–target interactions in animals and herbaceous plants, using various bioinformatics and molecular approaches. Conventional approaches for estimating miRNA–target interactions examine target specificity, such as the complementarity of the seed (2–7 nucleotides of the 5′ region of the miRNA) and the target (Brennecke et al. 2005). Transgenic approaches can also be used to elucidate miRNA–mRNA interactions, for example, by transforming with a target mRNA that has a mutated miRNA target site sequence. Lai et al. (2005) used this approach to reveal that miRNAs targeting genes from the Bearded and basic helix–loop–helix repressor families regulate many developmental processes in Drosophila. Choi et al. (2007) also used target protectors, which bind to the target site and disrupt the miRNA–mRNA interaction, to uncover the crucial role of miR-430-Squint (Nodal agonist) and miR-430-Lefty (Nodal antagonist) in enhancing or reducing Nodal signaling during zebrafish mesendoderm development, revealing the importance of specific miRNA–mRNA interactions in animal development. A series of studies in herbaceous plants have revealed the crucial roles of miRNA–target interactions, which affect all stages of development and produce phenotypic variation in many traits, such as leaf shape, flowering time, stature, and lateral root number (Aukerman and Sakai 2003; Palatnik et al. 2003; Chen 2004; Schwab et al. 2005).
Recently, several studies have used reverse genetics to examine the function of miRNA–mRNA interactions associated with wood formation (Du et al. 2011; Robischon et al. 2011; Lu et al. 2013). For example, mutation of the miRNA target site in a transgene expressing the class III HD-ZIP gene popREVOLUTA in hybrid aspen (Populus tremula × P. alba) made the mRNA resistant to the repressor miR166 (Robischon et al. 2011). Another class III HD-ZIP gene, POPCORONA (PCN), was specifically repressed by an artificial version of miR166, resulting in abnormal lignification. By contrast, overexpression of the miRNA-resistant form of PCN resulted in reduced lignification in xylem and phloem fibers. These studies verified the importance of the negative regulatory relationship between miRNAs and their targets in trees. Lu et al. (2013) showed that the transgenic Populus trichocarpa lines expressing Ptr-miR397a exhibited a severe reduction in expression of laccase, a predicted target gene of Ptr-miR397a, and showed a large decrease (40 %) in lignin content (LC), revealing the effect of the Ptr-miR397a–laccase interaction on phenotypic variation (Lu et al. 2013).
Transgenic and reverse-genetic methods are not sufficient to determine specific phenotypic consequences of miRNA–target interactions in trees, since trees are large, have long generation times, and lack characterized mutants. Also, since each miRNA can target many genes, ruling out the effects of other miRNA–target interactions and the indirect effects of target–target interactions remains a challenge (Bartel 2004). Moreover, in addition to the difficulties inherent in gene transformation, evaluation of the phenotypic effects of most specific interactions has remained a challenge. Consequently, developing a new approach to examine the phenotypic effects of miRNA–target interactions will provide an important advance in elucidating the genetic architecture of phenotypes derived from miRNA–target interactions in tree species.
Linkage disequilibrium (LD)-based association mapping has significant potential for identifying the natural allelic variation responsible for a specific phenotype (Sexton et al. 2012). In particular, candidate gene-based SNP association has been used to identify alleles associated with growth and wood properties in several tree species, including conifers, eucalyptus, and Populus (Thumma et al. 2005; Beaulieu et al. 2011; Du et al. 2013). SNP-based association mapping provides another approach for annotation of gene function and identification of genetic regulatory networks. Previous studies on SNPs in human pri-miRNAs and pre-miRNAs showed that specific SNPs lead to reduced levels of the mature miRNA and translation suppression of target mRNAs (Calin et al. 2005; Duan et al. 2007; Jazdzewski et al. 2008). Thus, SNP-based association may be used to elucidate the function of the miRNA and mRNA, and to examine the crucial roles of their interactions in complex traits of perennial tree species. In the present study, we identified Pto-miR530a and its predicted target gene Pto-KNAT1 from P. tomentosa; to examine their interaction, we developed a method combining RT-qPCR expression profiling and association mapping, the first such study for a forest tree species. Pto-miR530a and Pto-KNAT1 showed opposite expression levels in different tissues, indicating that Pto-miR530a negatively regulates Pto-KNAT1. SNP-based associations showed that eleven unique SNPs from Pto-miR530a and Pto-KNAT1 associated with the growth and wood quality traits, and the miRNA and target gene SNPs associated with the same traits, indicating that the miRNA and its target affect the same phenotypes. Epistasis analysis uncovered a strong effect of the miRNA–target interaction on these phenotypes, explaining from 0.1 to 3.56 % of the variation in each trait. Here, we used association genetics based on miRNA–mRNA mutations to study the phenotypic variation affected by the miRNA and target, as well as the miRNA–target interactions; this approach provides a new strategy to examine the genetic architecture of traits involving miRNA–target interactions.
Materials and methods
Population and phenotype
An association population of 460 unrelated individuals of P. tomentosa, representing almost the entire climatic zone and the original provenance of P. tomentosa, was used for SNP association studies (Du et al. 2013). These trees were randomly sampled from 1047 individuals assembled from an area of 1 million km2 along the Yellow River (30–40°N, 105–125°E), and were grown in Guan Xian County, Shandong Province, China (36°23′N, 115°47′E) using a randomized complete block design with three clonal replications (Du et al. 2012). The association population was divided into three large climatic regions, northwestern, northeastern, and southern, based on a principal components analysis and isodata fuzzy clustering using sixteen meteorological factors (Huang 1992).
Phenotypic data: nine quantitative traits were measured from the 460 individuals, and the traits included: tree growth traits such as diameter at breast height (DBH), tree height (H), and stem volume (V), and wood property traits such as holocellulose content (HC), α-cellulose content, LC, fiber length (FL), fiber width (FW), and microfiber angle (MFA). The measurement of these phenotypic data was described in detail by Du et al. (2014).
Identification and isolation of Pto-miR530a and its target gene Pto-KNAT1
Identification and isolation of Pto-miR530a: the developing xylem of stem, tension wood, and opposite wood was collected by gently scraping the partially lignified tissues of the outside of the stem and branch of a 30-year-old P. tomentosa clone “LM50”. Next-generation sequencing was used to identify miRNAs differentially expressed among these three tissues. From these miRNAs, Pto-miR530a was selected as a candidate for further study, since it showed different expression levels in response to mechanical stress during wood formation based on our study results. Pto-miR530a showed similar expression to its P. trichocarpa homolog Ptc-miR530a, as detailed by Lu et al. (2008). The full-length sequence of the Pto-miR530a gene was obtained by BLAST against the P. tomentosa genome sequence to identify the pre-mature sequence and the 200–300 bp on each side of the pre-mature miRNA region. The homologous sequences of miR530a were obtained from miRBase (http://www.mirbase.org/index.shtml) and the alignment was performed with MEGA 5.0 (Tamura et al. 2011).
Identification and isolation of Pto-KNAT1 and phylogenetic analysis: psRNATarget, a plant small RNA target analysis server (http://plantgrn.noble.org/psRNATarget/) was used to predict the targets of Pto-miR530a; this prediction produced 17 candidate targets. Comparison of the predicted targets of Pto-miR530a and the stem cambium cDNA library identified target genes that were involved in wood formation. Random end-sequencing of 1000 cDNA clones and comparison with Pto-miR530a sequences identified a full-length cDNA with high similarity to AtKNAT1 (82 % DNA sequence identity) as the candidate target of Pto-miR530a.
Degradome sequencing, which uses high-throughput sequencing to determine the sequence of RNA ends, has been widely and reliably used to identify microRNA (miRNA) cleavage sites (Addo-Quaye et al. 2008). Here, degradome sequencing of nine pooled tissues, including leaves, shoot apex, mature xylem, and developing xylem sampled from P. tomentosa, was performed to evaluate the regulatory relationship of the miRNA and the mRNA. To study the phylogenetic relationship of Pto-KNAT1 with the KNATs from other species, the amino acid sequences of KNAT were identified by searching public databases available from The Arabidopsis Information Resource (TAIR; http://www.arabidopsis.org/index.jsp), the TIGR rice genome annotation project (http://rice.plantbiology.msu.edu/), the Joint Genome Institute (JGI http://jgi.doe.gov/), and the maize genome (http://bioinfo.cau.edu.cn/ProFITS). The evolutionary distances were calculated using the Poisson correction method (Zuckerkandl and Pauling 1965). Multiple amino acid sequence alignment was carried out using MEGA 5.0 following the method described by Du et al. (2013). The evolutionary history was inferred using the Neighbor-Joining method (Saitou and Nei 1987).
Expression of Pto-miR530a and its target Pto-KNAT1 using reverse transcription quantitative PCR (RT-qPCR)
RT-qPCR was performed on a 7500 Fast Real-Time PCR System using the SYBR Premix Ex Taq as described in the manufacturer’s instructions. The cDNA template for the reactions was reverse-transcribed using total RNA extracted from root, shoot apex, cambium, developing xylem, mature xylem, phloem, mature leaf, and young leaf. Primer Express 3.0 software (Applied Biosystems) was used to design the primers for Pto-KNAT1. Poplar Actin (Accession Number: EF145577) was used as the internal control. The relative expression level of Pto-miR530a was measured as described by Song et al. (2013). Triplicate technical and triplicate biological repetitions were performed on all reactions. The results obtained from each tissue were standardized to Actin. Every reaction contained 2 µL of diluted cDNA (5 ng of total RNA), 10 µL of 2 × SYBR green PCR master mix (2×, Applied Biosystems) and 0.2 µmol each of the forward and the reverse primers, 0.4 µL ROX Reference Dye (50×) in a final volume of 20 µL. The conditions for PCR amplification were: 95 °C for 30 s, then 40 cycles at 95 °C for 3 s and 60 °C for 30 s for RT-qPCR amplification, and 1 cycle at 95 °C for 15 s, 60 °C for 1 min, and 95 °C for 15 s for dissociation.
SNP identification and genotyping
To find SNPs in Pto-miR530a and Pto-KNAT1, these genes were sequenced from 40 individuals randomly selected from the association population. Primers were designed based on cDNA sequences and amplified from these 40 individuals. The PCR products were recovered by agarose gel electrophoresis and purified using Ultra free H-DA centrifugal filter units (Millipore, Billerica, MA, USA). The purified PCR products were then ligated into the pEASY-T1 Simple Cloning Vector (TransGen Biotech, Beijing, China) and transformed into BMTOP10 competent cells (Promega) according to the manufacturer’s protocol. Plasmid DNA was extracted and sequenced using the methods described by Tian et al. (2012). Sequences of these two genes from the 40 individuals were analyzed and SNPs were detected using MEGA 4.0. Thirteen common SNPs (minor allele frequency >10 %) from Pto-miR530a, and 29 common SNPs from Pto-KNAT1 were genotyped in 460 individuals of the association population using the method described by Du et al. (2013).
Data analysis
Multiple SNP association The fGWAS Package (Functional Genome-wide Association Studies) for simultaneous analysis and genetic modeling of multiple SNPs was used to identify and estimate the possible additive and dominant effects associated with all significant SNPs for each trait, and was run in R (http://www.r-project.org/). This package analyzes the genotypic and phenotypic data through a preconditioning step that mitigates the observational noise with a supervised principal component analysis (PCA) and variable selection step with an implemented Bayesian Lasso model, and then summarizes the significant SNPs at a significance level of 0.01 (Li et al. 2010).
Epistasis analysis Epistasis, the nonlinear interactions between DNA variants, can be used to identify the polymorphic loci for quantitative traits and expose the genetic architecture associated with these traits. Multifactor Dimensionality Reduction 3.0.2 (MDR 3.0.2) was applied to investigate epistatic interactions in our study (Jakulin and Bratko 2003; Jakulin et al. 2003). The MDR algorithm detects interactions by combining attribute selection, attribute construction, classification, and permutation testing. In the MDR software, the ReliefF algorithm (to improve the reliability of the probability approximation) was employed to produce the five most informative SNPs, and tenfold cross-validation was used to generate all possible combinations of two to five SNPs at a significance level of 0.001 (Robnik-Siknja and Kononenko 2003). The information gain was calculated by an entropy-based measure to evaluate the genetic effects of significant SNP–SNP interactions.
Results
Identification of Pto-miR530a and Pto-KNAT1
Identification of Pto-miR530a Next-generation sequencing results for total miRNAs from three different tissues showed that Pto-miR530a is abundant in tension wood (5.59-fold more than in stem) and opposite wood (4.8-fold more), indicating that Pto-miR530a may be involved in wood development associated with mechanical stress. The sequencing results yielded the 20-nt sequence of the mature miRNA of Pto-miR530a (5′-UGCAUUUGCACCUGCACCUU-3′, accession number KP058661) and BLAST against the P. tomentosa genome yielded the 144-nt sequence of the pre-mature miRNA (KP058662) and 679 bp of genomic sequence including 200–300 bp on each side of the pre-mature miRNA (KP058620). Prediction of the secondary structure of Pto-miR530a using RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) analysis of the pre-mature miRNA sequence revealed a typical hairpin structure, confirming that Pto-miR530a is a miRNA (Fig. S1). Alignment of Pto-miR530a with the homologous miRNAs from other species, including P. trichocarpa, rice, Citrus sinensis, Theobroma cacao, and Glycine max, showed that despite the high nucleotide diversity of pre-mature regions, the seed regions of these miR530s are highly conserved.
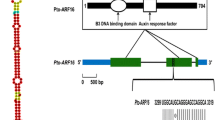
Identification of Pto-KNAT1 The 20-nt mature sequence of Pto-miR530a was used to predict its targets, using psRNA Target; this identified 17 candidate mRNAs. Comparison of the candidate mRNAs and sequenced cDNAs from the stem cambium cDNA library identified Pto-KNAT1 as one of the predicted targets of Pto-miR530a and indicated that it binds Pto-KNAT1 at 229–249 nt (Fig. 1a). To further examine the regulatory relationship between Pto-miR530a and Pto-KNAT1, we performed degradome sequencing, which showed that the most likely cleavage site was at 240 nt, indicating that Pto-miR530a regulates Pto-KNAT1 by cleaving its 5′-untranslated region (5′-UTR, Fig. 1b). The full-length Pto-KNAT1 cDNA was isolated by reverse transcription PCR from the cDNA library constructed from the cambium zone of P. tomentosa. The cDNA of Pto-KNAT1 is 2029 bp, with a coding region of 1107 bp encoding 368 amino acids, flanked by a 595-bp 5′-UTR and a 307-bp 3′-UTR (KP058660). Sequence analysis of Pto-KNAT1 cDNAs showed that Pto-KNAT1 contains four conserved regions, including a KNOX 1 domain (106–149), KNOX 2 domain (158–207), ELK domain (250–271), and homeobox domain (295–330) (Fig. 2).

Possible cleavage sites of Pto-KNAT1 identified by degradome sequencing. a Pairwise alignment of Pto-miR530a and Pto-KNAT1 performed by psRNATargets (http://plantgrn.noble.org/psRNATarget). b Possible cleavage sites of Pto-KNAT1 identified by degradome sequencing

Structure of Pto-KNAT1. Pto-KNAT1 consists of a 602-bp promoter, 238 bp of 5′ untranslated region (5′-UTR), 1107 bp of exon (five exons: 390, 123, 136, 254, and 204 bp), 4371 bp of intron (four introns: 118, 191, 3908, and 155 bp), and 307 bp of 3-UTR. The coding sequence of Pto-KNAT1 contains four important domains: KNOX 1, KNOX 2, ELK, and homeobox domains, which characterize Pto-KNAT1 as a class I KNOX gene family member
To examine the evolutionary relationship of Pto-KNAT1 with KNATs from other species including Arabidopsis thaliana, Oryza sativa, and Zea mays, we used MEGA 5.0 to generate an unrooted tree from 24 protein sequences (Fig. 3). The phylogenetic tree showed two subcategories, representing class I and class II of the Knotted-like family, indicating that the gene duplication event that gave rise to the two subcategories occurred before the divergence of the monocot and dicot lineages. Phylogenetic analysis revealed that Pto-KNAT1 has close homology with Pt-KNAT1.1 (Potri.002G113300.1), indicating that Pto-KNAT1 belongs to class I in the Knotted-like gene family.

A phylogenetic tree of homologs of Pto-KNAT1 from Arabidopsis thaliana, rice (Oryza sativa), and maize (Zea mays). The evolutionary history was inferred using the Neighbor-Joining method. Protein sequences of Arabidopsis KNAT family members (KNAT1-7, STM) were obtained from The Arabidopsis Information Resource (TAIR; http://www.arabidopsis.org/index.jsp) and rice members (LOC) were obtained from the TIGR rice genome annotation project (http://rice.plantbiology.msu.edu/). Maize (GRMZM) family members were obtained from the maize genome (http://bioinfo.cau.edu.cn/ProFITS/gene_brw.php?src=4a53&gene_id=GRMZM2G094241)
Tissue-specific expression of Pto-miR530a and Pto-KNAT1
To determine whether Pto-miR530a and Pto-KNAT1 show tissue-specific expression, we used RT-qPCR to measure their expression levels in different tissues. Pto-miR530a and Pto-KNAT1 transcripts occurred in all organs in different abundances (Fig. 4). We observed a higher level of Pto-miR530a in young leaves, followed by mature leaves, and we observed a lower level in mature xylem, followed by shoot apex. By contrast, we observed the highest transcript level of Pto-KNAT1 in mature xylem (17.28), followed by developing xylem (8.72), indicating that Pto-KNAT1 may be involved in secondary wood formation. Also, we observed low levels of Pto-KNAT1 transcript in young leaves (0.23) and mature leaves (0.67), and moderate levels in cambium (4.46), indicating that Pto-KNAT1 may also be involved in primary wood formation. Thus, the abundance of Pto-miR530a and Pto-KNAT1 showed opposite patterns. Their Pearson’s product–moment correlation was r = −0.62 (P < 0.05), indicating strong correlation of transcription levels between Pto-miR530a and Pto-KNAT1.

Expression of Pto-miR530a and Pto-KNAT1 in different tissues. mRNA and miRNA levels were measured in eight tissues: young leaves, mature leaves, phloem, cambium, root, shoot apex, developing xylem, and mature xylem, by RT-PCR. Actin was used as the internal control
Nucleotide diversity and linkage disequilibrium in Pto-miR530a and Pto-KNAT1
To perform multi-SNP association mapping, we used genomic re-sequencing to identify SNPs in Pto-miR530a and Pto-KNAT1. To measure the intraspecific nucleotide diversity, we obtained approximately 679 bp and 6788 bp of genomic sequence for Pto-miR530a and Pto-KNAT1, respectively, by direct sequencing of 40 unrelated individuals of P. tomentosa. The 40 full sequences of Pto-miR530a and Pto-KNAT1 were deposited in NCBI (http://www.ncbi.nlm.nih.gov/) under the accession numbers KP058620–KP058659 and KP058580–KP058619, respectively. Comparison of the 40 individual sequences identified 40 SNPs in Pto-miR530a and 268 SNPs in Pto-KNAT1, a frequency of 1/17 bp (π T = 0.01435 and θ w = 0.01444) and 1/25 bp (π T = 0.01004 and θ w = 0.0094), respectively (Table 1). The most conserved region was the mature region of Pto-miR530a, in which no SNPs were identified, as expected since mature miRNAs are extremely well conserved among species. The exon regions in target genes were also conserved, consistent with the expectation that the coding regions are under stronger selection pressure. The most abundant nucleotide polymorphism was observed in the promoter region of Pto-KNAT1 (π T = 0.02009 and θ w = 0.02172). Eighteen SNPs were detected in exons, including seven nonsynonymous mutations and eleven synonymous mutations. Also, 46.6 % of the 268 total SNPs were common SNPs (frequency >0.10). We observed one common SNP (Pto-KNAT1-8462166) in the first nucleotide of the Pto-miR530a binding site of Pto-KNAT1. To identify the effect of this single SNP, we used psRNATarget to calculate the target accessibility (allowed maximum energy to unpair the target site, UPE) and maximum expectation (E) for A/G transition. The maximum expectation changed from 4.0 (A) to 4.5 (G), and required more energy to unpair the target site, indicating that the A allele of Pto-KNAT1 produces a stronger effect on cleavage of the Pto-KNAT1 mRNA by Pto-miR530a, compared with the G allele, and suggesting that the two alleles may differ in their phenotypes (Table S1).
For the association analysis, we selected 42 common SNPs (minor allele frequencies >0.10) from Pto-miR530a and Pto-KNAT1 and genotyped these SNPs in 460 unrelated individuals in the association population, using the method described by Du et al. (2013). The patterns of linkage disequilibrium (LD) decay of Pto-miR530a and Pto-KNAT1 were calculated to evaluate whether the two genes are suitable for candidate gene-based association analysis. LD declined rapidly within 600 bp in Pto-miR530a (R 2 < 0.1, P < 0.001, Fig. 5) and within 1400 bp in Pto-KNAT1 (R 2 < 0.1, P < 0.001), which means that the LD of both genes did not extend to the total gene sequence. Therefore, Pto-miR530a and Pto-KNAT1 are suitable for candidate gene-based association analysis.

Linkage disequilibrium of Pto-miR530a (a) and Pto-KNAT1 (b) from three different geographical regions and the entire region. NE northeast region, NW northwest region, S south region; total, samples from northeast, northwest, and south regions analyzed as a whole
Multiple SNP-based associations with additive and dominant effects
Our examination of the expression of Pto-miR530a and Pto-KNAT1 in different tissues showed that they are closely associated with wood formation. Therefore, we used a Bayesian Lasso model employing a Markov chain Monte Carlo algorithm to estimate the additive and dominant effects of individual SNPs for each quantitative trait, also considering the effects of all other SNPs and covariates. The algorithm simultaneously identified multiple SNPs associated with the phenotypic variance and calculated the R 2 of every SNP, finding 37 associations (P < 0.01) with 9 traits representing tree growth and wood properties. Of these associations, 86.5 % showed additive effects (Table 2), 91.9 % showed dominant effects (Table 3), and 73.0 % showed a combination of additive and dominant effects (Table 4).
We identified 32 significant associations with additive effects; these associations involved 11 unique SNPs and 9 traits. The numbers of detected SNP–trait associations differed for different trait categories, with 10 significant associations for tree growth traits, 10 for wood physical properties, and 12 for wood chemical composition traits. The additive effects of each SNP ranged from 0.16 to 18.09 (Table 2). Among these, Pto-KNAT1-8462663, which affects the Pto-KNAT1 coding region and associated with DBH, had the largest effect (18.09), and Pto-KNAT1-8502875, which is in a noncoding region and associated with LC, had the smallest effect (0.16). Of the associations with additive effects, 74.1 % were associated with wood properties, with an average effect of 3.74. We also identified 34 significant associations with dominant effects; these associations involved 13 unique SNPs and 9 traits. The dominant effects of each SNP ranged from −14.08 to 19.00 (Table 3). Of these, 81.5 % were positive dominant values and 18.5 % were negative. Pto-miR530a-1360249, which was associated with α-cellulose, had the largest positive effect (19.00), and Pto-KNAT1-8462213 (in the 5′-UTR), which was associated with DBH, had the largest negative effect (−14.08). Each trait was associated with 2–8 SNPs. For example, α-cellulose was associated with 8 SNPs, 5 from Pto-KNAT1 with dominant effects ranging from −3.09 to 0.74, and 3 from Pto-miR530a with dominant effects ranging from 6.00 to 19.00.
In total, 27 associations were identified across 8 traits representing tree growth and wood properties; these had a combination of additive and dominant effects, with the R 2 of each SNP ranging from 0.83 to 15.81 % (Table 4). We constructed a hierarchical network based on all 27 associations between 11 unique SNPs and 8 tree growth and wood property traits (Fig. 6). Of the eleven significant SNPs identified by this model, five SNPs were from Pto-miR530a and six SNPs were from Pto-KNAT1. 45.6 % of these SNPs were associated with 2–6 traits, which likely can be attributed to the strong correlation between tree growth and wood property traits. Interestingly, for eight traits, significant SNPs in Pto-miR530a and Pto-KNAT1 both contributed to the same trait. For example, the SNPs Pto-KNAT1-8462166 and Pto-KNAT1-8462338 from Pto-KNAT1 and Pto-miR530a-1359910, Pto-miR530a-1359964, and Pto-miR530a-1360249 from Pto-miR530a were significantly associated with α-cellulose, with R 2 values that ranged from 0.83 to 15.02 %. Also, Pto-KNAT1-8462166, Pto-KNAT1-8462338, and Pto-miR530a-1359964 were associated with HC, with the R 2 of each SNP ranging from 0.83 to 4.68 %. Interestingly, the identified SNPs showed different contributions to the same traits. For example, Pto-miR530a-1360287, Pto-miR530a-1360318, and Pto-miR530a-1359964 were associated with H, with the R 2 explained by each SNP ranging from 1.58 to 15.81 %, while Pto-KNAT1-8462166 contributed to the same trait with an R 2 of 1.1 %, which was much lower than that of the SNPs from the miRNA. The same situation was observed for DBH, α-cellulose content, and LC, where the variance explained by SNPs in Pto-miR530a ranged from 13.33 to 15.02 %, but the variance explained by SNPs in Pto-KNAT1 only ranged from 0.82 to 3.42 %. However, SNPs of Pto-KNAT1 explained more of the variance in HC, FL, and FW at 4.68, 6.86, and 1.09 %, respectively, much more than the variance explained by SNPs in Pto-miR530a, at 1.66, 0.87, and 0.84 %, respectively. Thereafter, we calculated genetic variances explained by significant SNPs using the mixed linear model in TASSEL (Table S2). Of the 27 significant SNPs detected by the Bayesian Lasso model, 70.4 % of them were also significant in the mixed linear model. No significant difference was observed between the average R 2 explained by fGWAS (average 6.97 %) and by the mixed linear model (6.64 %).

The phenotypic variance (R 2) of SNPs in Pto-miR530a and Pto-KNAT1 across eight tree growth and wood property traits calculated by multi-SNP associations. MFA microfiber angle, FW fiber length, FL fiber width
How do the genotypic effects manifest when more than two significant SNPs contribute to the same trait? Taking α-cellulose as an example, three loci from Pto-miR530a (Pto-miR530a-1359910, Pto-miR530a-1359964 and Pto-miR530a-1360249) and two loci from Pto-KNAT1 (Pto-KNAT1-8462166 and Pto-KNAT1-8462338) contribute to this trait. The three genotypes of each locus had different effects on the phenotype (Table 5). For example, the AG heterozygotes of Pto-miR530a-1359910 were associated with higher α-cellulose values (40.00 %), followed by GG (39.94 %), and AA homozygotes were associated with the lowest α-cellulose values (35.81 %). Simultaneously, in locus Pto-KNAT1-8462166, higher α-cellulose associated with the AA homozygotes (40.72 %), followed by AG heterozygotes (39.89 %), and GG homozygotes were associated with lowest α-cellulose values (34.66 %), consistent with the multi-SNP association results. When we consider all the loci, the AG-CC-TG-AG-AT genotype was associated with the highest α-cellulose content (50.06 %), followed by the AA–AA-TG-AG-AA genotype (44.45 %), and the AG-AC-TG-GG-AA genotype was associated with the lowest α-cellulose content (26.4 %) (Table 6).
Epistasis between SNPs in Pto-miR530a and Pto-KNAT1
Besides additive and dominant effects, epistasis also contributes to phenotypic variance. We used MDR 3.0.2, a novel algorithm that can identify SNP–SNP interactions in a population, to calculate the epistatic interactions of SNPs in Pto-miR530a and Pto-KNAT1. MDR and the ReliefF algorithm reliably identified the five most informative SNPs for each trait and calculated the independent information gain (IG) to measure the contribution of each SNP. After statistical significance analysis, 65 associations (Q ≤ 0.10) were identified with 8 traits, including 23 unique SNPs from 2 genes, with main effects ranging from 0.02 to 2.75 % (Table S3). Of the 23 unique SNPs, three SNPs also showed significant additive and dominance effects across six traits. By analyzing the pairwise effects, 65 SNP pairs were identified with epistatic interactions ranging from 0.10 to 3.56 %. Of the total SNP–SNP interactions, 36.9 % represented miRNA–mRNA interactions and 63.1 % represented miRNA–miRNA or mRNA–mRNA interactions. The SNP pairs formed a highly interconnected network representing the epistatic interactions between these polymorphic loci, which reflect the interaction of Pto-miR530a and Pto-KNAT1 (Fig. 7).

The epistatic interactions of different loci from Pto-miR530a and Pto-KNAT1. Different colors represent the results generated by different phenotypic traits. The solid lines indicate that the two loci together have larger effects on the trait than the sum of the two individual effects; the dashed lines indicate that the two loci together have a smaller effect than the sum of the two individual effects. FL fiber length, H height, FW fiber width, DBH diameter at breast height, MFA microfiber angle, CC α-cellulose content, HC holocellulose content, V volume. SNP1 Pto-KNAT1-8461839, SNP2 Pto-KNAT1-8461939, SNP4 Pto-KNAT1-8462166, SNP5 Pto-KNAT1-8462213, SNP7 Pto-KNAT1-8462416, SNP10 Pto-KNAT1-8463273, SNP11 Pto-KNAT1-8463888, SNP13 Pto-KNAT1-8464801, SNP15 Pto-KNAT1-8467424, SNP16 Pto-KNAT1-8469178, SNP17 Pto-KNAT1-8471625, SNP18 Pto-KNAT1-8474222, SNP22 Pto-KNAT1-8486299, SNP24 Pto-KNAT1-8494130, SNP27 Pto-KNAT1-8508341, SNP29 Pto-KNAT1-8514062
The epistatic effects of SNP pairs were evaluated by analysis of information gain (IG), which was implemented in the MDR algorithm. The results showed that the genetic effects of significant SNP pairs ranged from −3.09 to 0.93 % (Table S4). Of the total IGs for the SNP–SNP interactions, 95.4 % were negative. For example, for the H trait, 10 significant SNP pairs involving 5 unique SNPs were observed with the IG ranging from −1.31 to 0.93 %. Pto-KNAT1-8471625 and Pto-KNAT1-84619397 have nonlinear interactions (IG = 0.93 %), followed by Pto-KNAT1-8461939/Pto-KNAT1-8471625 (IG = 0.56 %), and Pto-miR530a-1360155/Pto-KNAT1-8471625 (IG = 0.45 %), indicating that these three SNP–SNP interactions explained more phenotypic variance than the single SNPs. However, most of the IGs were negative. For example, IGs of Pto-KNAT1-8467424/Pto-KNAT1-8467584, Pto-KNAT1-8467424/Pto-miR530a-1360155, and Pto-KNAT1-8461939/Pto-KNAT1-8471625 were −0.31, −0.98, and −0.96 %, respectively.
Transcript analysis in significant SNP genotypes
To determine whether Pto-KNAT1 expression levels differed in the different genotypic classes for the miRNA and mRNA, we used RT-qPCR with gene-specific primers to compare transcript levels of Pto-KNAT1 in different trees with eight different genotypes (the T:T/C:C genotype did not occur in the association population). The experiments used secondary xylem from 30-year-old trees to measure Pto-KNAT1 mRNA levels in 24 trees (3 trees for each genotype pair) of the association population. The transcript levels of Pto-KNAT1 varied in the different genotype pairs (Fig. 8). When the genotype of Pto-miR530a-1359964 was C:C or A:C, the transcript levels of three different genotypes (A:A, A:T and T:T) in the locus Pto-KNAT1-8474222 did not show a significant difference (P < 0.01); however, when the genotype was T:T, the three genotypes of Pto-KNAT1-8474222 did show significant differences. For the genotype A:A in Pto-miR530a-1359964, the highest transcript level was observed in the trees where Pto-KNAT1-8474222 was A:T, followed by C:C, and A:A, respectively.

Transcript levels of Pto-KNAT1 in eight genotypic classes for Pto-miR530a-1359964 and Pto-KNAT1-8474222. Three genotypes (A:A, A:T, and T:T) of locus Pto-miR530a-1359964 were observed; three genotypes (A:A, A:C, and C:C) of locus Pto-KNAT1-8474222 were observed. For each genotype pair, three trees were selected and their transcript levels were measured. The genotype pair of T:T/C:C was not observed. *P < 0.05; **P < 0.01
Discussion
Many genes encode proteins that activate or repress the transcription of other genes through complex interactions. Especially for long-lived species, multiple minor genes control most traits, such as tree growth and wood properties. These genes and their interactions form a genetic regulatory network to maintain the body plan and environmental responses of these highly organized, complex organisms. In this network, focusing on the identification and functional annotation of miRNAs and on miRNA–mRNA interactions can improve our understanding of the mechanisms of gene regulation and phenotypic variance in trees (Lu et al. 2005, 2013). The characterization of miRNA–mRNA interactions will provide us with new insights into the mechanisms of tree growth and wood formation.
Association is essential for fine-mapping of candidate genes and construction of genetic networks in forest trees
Reverse genetics-based annotation of gene function in long-lived forest trees is difficult due to their large genomes, complex regulatory networks, and adaptation to environmental changes. SNP-based association mapping provides more reliable marker–trait associations in natural populations than family-based quantitative trait locus studies. Association mapping has been widely used to identify significant marker–trait associations, which provide an additional method for annotation of gene function (Thornsberry et al. 2001; Thumma et al. 2005). Therefore, studying the function of SNPs also allows us to study the function of genes. Similarly, studying SNP–SNP interactions allows us to study the interaction of the two candidate genes that harbor the SNPs. Understanding the interactions between SNPs can be used to infer the genetic regulatory network affecting complex traits, significantly improving our understanding of the underlying biological process (Phillips 2008).
Association studies must consider the extent of LD. In the present study, we observed a rapid decay of LD within 600 bp in Pto-miR530a and within 1,400 bp in Pto-KNAT1 (R 2 < 0.1, P < 0.001, Fig. 5a, b), which is much lower than that observed in P. trichocarpa with a slower LD decay of more than 3 kb at R 2 = 0.2 (Zhou et al. 2014). We found that the LDs reported in these two recent papers decayed within 100 kb, which is much longer than the distances observed in our study. We think that one of the main reasons for this would be that the LD in our paper was calculated based on nucleotides within candidate genes, which may limit the distance of the nucleotides, leading to rapid decay of the LD in these genes. This is a technology-limited problem, which we are trying to conquer through deep sequencing. Another reason is that an outcrossing species, a large effective population size, and a long history of recombination, may lead to the rapid decay of LD. For example, in maize (an outcrossing species) and some forest tree species, rapid decline in LD (less than 1 kb) was observed in many candidate genes (Thornsberry et al. 2001; Thumma et al. 2005).
Different association methods may emphasize different issues; thus, it is important to select an appropriate statistical method. The fGWAS approach involves a two-stage procedure for multi-SNP modeling and analysis in GWAS, using supervised PCA and then formulating a Bayesian lasso to select a subset of significant SNPs (Li et al. 2010). The mixed models are especially useful in settings where repeated measurements are made on the same statistical units, or where measurements are made on clusters of related statistical units. Here, we calculated genetic variances explained by significant SNPs using a mixed linear model in TASSEL (Table S2). Our results showed that 70.4 % of the 27 significant SNPs detected by the Bayesian Lasso model were also detected in the mixed linear model, and we found no significant difference between the average R 2 explained by the two models. This suggests that population structure played an insignificant role in our study, which is consistent with previous reports that tree species are predominantly outcrossing and have large, relatively unstructured populations (Thumma et al. 2005). Ultimately, fGWAS is a powerful tool for identification of multiple SNPs associated with complex traits with dominant and additive effects.
In the association study based on multiple SNPs, we found that SNPs in Pto-KNAT1 were associated with both tree growth (H and DBH) and wood properties, which is gratifying, since selection for improvement of crop trees generally considers both traits concurrently. In total, we detected 13 significant associations between SNPs in Pto-KNAT1 and traits (Table 4); by contrast, work in other species detected many fewer associations (Thumma et al. 2005; Southerton et al. 2010). This may be because we used more phenotypes to identify significant associations. Several SNPs were associated with different traits, which may reflect the diverse functions of Pto-KNAT1 in wood properties and tree growth, and is in accordance with a previous study showing that the homologs of Pto-KNAT1 function in xylem and phloem pattern formation, xylem development (Du et al. 2009; Liebsch et al. 2014), and shoot apical meristem maintenance (Scofield and Murray 2006). For example, transcript levels of six genes involved in cellulose synthesis and six genes involved in lignin biosynthesis were negatively correlated with expression of ARK2, a homolog of Pto-KNAT1 in P. trichocarpa, indicating that these wood property genes may be regulated by ARK2 (Du et al. 2009; Liu et al. 2015). Furthermore, transgenic lines overexpressing and suppressing ARK2 showed opposite expression levels of several key transcriptional regulators, such as class III HD-ZIP transcription factors and WUSCHEL, and genes involved in cell division, cell expansion, and hormone responses. Pto-KNAT1 affects many growth and wood properties suggesting that, as a transcription factor, Pto-KNAT1 may regulate many downstream genes (Du et al. 2009; Liu et al. 2015). We were surprised to find that some SNPs were significantly associated with more than one trait, such as Pto-KNAT1-8462166 and Pto-KNAT1-8462338, but others were associated with only one trait, such as Pto-KNAT1-8502875 and Pto-KNAT1-8508341. This may be due to several reasons: first, Pto-KNAT1-8462166, Pto-KNAT1-8462338, and Pto-KNAT1-8462213 are located in the 5′ UTR of Pto-KNAT1. The allelic variance in this region may affect Pto-KNAT1 transcript levels, since the 5′ UTR is an important regulatory region that is bound by many activators and repressors (Southam et al. 2007; Tian et al. 2012). Second, SNPs in the exonic regions may affect codon usage and transcript abundance (Kimchi-Sarfati et al. 2007; Chamary and Hurst 2009); for example, Pto-KNAT1-8462663 causes a synonymous substitution and was associated with DBH. Third, significant SNPs located in introns, such as Pto-KNAT1-8502875 and Pto-KNAT1-8508341, may affect phenotypic traits by regulating gene expression and exon splicing (Tian et al. 2012). The SNPs of Pto-KNAT1, such as Pto-KNAT-84508341, Pto-KNAT1-8502875, Pto-KNAT1-8462213, and Pto-KNAT1-8462663, exclusively affect microfiber angle, lignin content, and DBH, indicating that Pto-KNAT1 plays important roles in several pathways, including carbohydrate and lignin pathways (Sexton et al. 2012).
A previous study of miRNAs in P. trichocarpa identified Ptc-miR530a, a homolog of Pto-miR530a. The mature sequences, as well as the pre-mature sequences of these two miRNAs, match perfectly, indicating that these miRNAs are highly conserved between these two species. Surprisingly, unlike Ptc-miR530a, whose transcript levels were significantly higher, by more than 1.5-fold, in tension wood than that in opposite wood, Pto-miR530a does not show differential expression in these two tissues. This suggests that the members of these miR530a may have different functions in these two tree species, even though their sequences are highly conserved (Lu et al. 2008).
Target recognition revealed the interaction of Pto-miR530a with Pto-KNAT1
With the development of diverse prediction methods, thousands of miRNA/mRNA interactions have been predicted, but few have been experimentally verified. However, requiring conserved Watson–Crick pairing in the seed region of the miRNA significantly decreases the false-positive predictions, thus substantially improving the identification of miRNA targets (Lewis et al. 2003, 2005). This conclusion agrees with previous studies that the 5′ region is the most conserved portion of miRNAs and pairs with perfect complementarity to sequences in the 3′-UTR that regulate mRNA degradation and translational repression in some metazoans (Lim et al. 2003; Lai et al. 2005). Therefore, the seed region is sufficient for prediction of miRNA targets (Krek et al. 2005). This has been widely used in mammalian and plant target prediction algorithms that simply search for mRNAs with conserved nucleotides complementary to 2–7 nt of the seed region (Lewis et al. 2005); however, algorithms using 8 nt matches to the miRNA showed higher confidence predictions. In our study, a 20-nt miRNA, Pto-miR530a, with sequence 5′-UGCAUUUGCACCUGCACCUU, was identified in P. tomentosa. The 6-nt-conserved seed region GCAUUU of Pto-miR530a matches the 5′-UTR of Pto-KNAT1 with the sequence of CUUAAA, with an expectation of 4.5 and target accessibility of 12.801, indicating a modest confidence of target recognition. The 8-nt sequence of UGCAUUUG was also considered to increase the confidence of target specificity.
The imperfect match of the seed region to the miRNA raises the question of whether Pto-KNAT1 is the target of Pto-miR530a. To address this question, we examined the expression of Pto-miR530a and Pto-KNAT1 in different tissues, as gene expression analysis can help identify the targets of miRNAs (Palatnik et al. 2003). We detected Pto-miR530a and Pto-KNAT1 mRNA in every tissue, in agreement with previous reports that targets with imperfectly matched seed sites are expressed in the same tissues as their targeting miRNAs (Farh et al. 2005). Many more targets with imperfect matches than with perfect matches to their targets were identified after the endogenous miRNA was deleted or inhibited (Selbach et al. 2008). Another study showed that a large portion of messages with imperfectly matched sites still have functions even when the miRNA is expressed in the same cell (Farh et al. 2005). These studies indicated that Pto-KNAT1 may be a target of Pto-miR530a, although it contains an imperfectly matched seed site. Further analysis showed a negative correlation of the expression levels of Pto-miR530a and Pto-KNAT1 (Fig. 4), indicating that the miRNA affects the levels of the mRNA (Baek et al. 2008; Lu et al. 2013). This conclusion is consistent with a previous genome-wide expression profiling of human populations of European and African ancestry, which showed that increased levels of miRNAs are related to consistently reduced transcript levels of their target mRNAs (Gamazon et al. 2012).
Multi-SNP association reveals the miRNA–mRNA interaction
Although single SNP-based GWAS has been widely used to detect important genes associated with numerous diseases and traits, GWAS has several shortcomings that limit its application (Donnelly 2008). For example, these strategies only can detect a small percentage of genetic variation and cannot identify weaker SNPs; they also cannot identify the interactions of multiple genes associated with complex traits (Li et al. 2010). The fGWAS strategy, which can identify many SNPs associated with one trait, reflects the genetic network formed by different interactions (Li et al. 2010). Our study used fGWAS to study multiple SNPs affecting the same traits, and to study the interaction of the identified SNPs. The significant SNPs from Pto-miR530a and Pto-KNAT1, including five SNPs from Pto-miR530a and six SNPs from Pto-KNAT1, formed a hierarchical regulatory network affecting eight traits (Fig. 5). Five of the SNPs were associated with 2–6 traits, for example, Pto-KNAT1-8462338 associated with both wood chemical properties such as α-cellulose, HC, and LC and with wood physical properties such as FW and MFA, indicating pleiotropic effects of these important loci (Tian et al. 2012). Similarly, each trait was affected by more than two SNPs. For example, three SNPs (Pto-KNAT1-8462213, Pto-KNAT1-8462663, and Pto-miR530a-1359964) were significantly associated with DBH, indicating that Pto-miR530a and Pto-KNAT1 may contribute to the same pathway in the radial growth of woody plants (Du et al. 2011). Significant SNPs in Pto-miR530a and Pto-KNAT1 both associated with the same trait, reflecting the possible genetic interaction of the miRNA and its target gene.
We observed that SNPs from Pto-miR530a and Pto-KNAT1 made different contributions to the same traits. For example, SNPs from the miRNA, such as Pto-miR530a-1360287, Pto-miR530a-1360318 and Pto-miR530a-1359964, were associated with H, with R 2 ranging from 1.58 to 15.81 %, and Pto-KNAT1-8462166 was associated with the same trait, with an R 2 of 1.1 %, which was much lower than the variance explained by SNPs from the miRNA, indicating that SNPs from the miRNA may have larger effects on tree growth. A similar situation was also found for wood chemical composition, such as α-cellulose content and LC, indicating the Pto-miR530a may be more important than the SNPs in Pto-KNAT1 for these traits (Lu et al. 2013). However, SNPs from Pto-KNAT1 explained a much higher variance for other traits, such as FL and FW, than SNPs from Pto-miR530a, indicating that Pto-KNAT1 is more important for wood physical properties (Du et al. 2009). These conclusions were further verified by RT-qPCR (Fig. 4), which showed that Pto-miR530a and Pto-KNAT1 were least and most abundant, respectively, in xylem, indicating that these genes may have functions in cell wall biochemistry and lignin composition (Groover et al. 2006), consistent with a potential effect on wood chemical and physical properties. Pto-miR530a had lower transcript levels in the shoot apex, indicating that this miRNA had less of a negative effect on mRNAs associated with tree growth traits (Du et al. 2011). We further analyzed the effects of Pto-miR530a-1359910, Pto-miR530a-1359964, Pto-miR530a-1360249, Pto-KNAT1-8462166, and Pto-KNAT1-8462338, which were significantly associated with α-cellulose. We found that the genotypes of different loci had diverse effects on phenotype and the results were consistent with the observation that these loci had additive and dominant effects on α-cellulose (Du et al. 2009, 2014). The genetic effects of the five loci, with genotypes AG-CC-TG-AG-AT and AG-AC-TG-GG-AA, were associated with the highest and lowest α-cellulose content, respectively, indicating that the combined effects of the loci from the miRNA and mRNA may have a larger effect than any single SNP. We found that AG-AC-TG-AA-AA was associated with higher α-cellulose content (44.24) than AG-AC-TG-AG-AA (38.64) and AG-AC-TG-GG-AA (26.4), indicating that the single SNP in the miRNA targeting site (Pto-KNAT1-8462166) in the 5′-UTR of Pto-KNAT1 could lead to significant phenotypic variance (Zhang et al. 2011). We noted that Pto-miR530a likely had a stronger ability to induce cleavage of Pto-KNAT1 transcripts with the A allele than those with the G allele, indicating that Pto-KNAT1 may negatively regulate genes associated with α-cellulose content.
Epistasis analysis reveals the miRNA–mRNA interaction
Epistasis was first devised as an explanation for deviation from simple Mendelian ratios (Bateson 1909) and examination of epistasis can provide insights into genetic pathways and biological mechanisms (Maher 2008; Natarajan et al. 2013a, b). The genetic architecture of common human diseases and the significant roles of epistasis in disease susceptibility indicate that complex genetic interactions may have stronger effects than any single susceptibility gene (Chou et al. 2011). Although molecular experiments have demonstrated epistatic interactions for qualitative traits (Babu et al. 2014), GWAS has identified few epistatic interactions for quantitative traits because of the great amount of work required to compute all pairs of SNPs (Natarajan et al. 2013a, b). MDR was developed to address this challenge and it provides a powerful tool for discovering nonlinear interactions of different genes or loci.
Our study used MDR to calculate SNP–SNP interactions, to investigate the interaction between Pto-miR530a and Pto-KNAT1. We found 65 SNP–SNP interactions, and 36.9 % of them contained SNPs located in Pto-miR530a and Pto-KNAT1, respectively. About 95.4 % of the total IGs for the different SNP pairs were negative, indicating that these mutant loci provide some redundant information for the same trait (Collins et al. 2013). Of the 24 miRNA–mRNA interactions, 95.8 % of their IGs were negative, suggesting that Pto-miR530a and Pto-KNAT1 share redundant information and may contribute to the same pathways in wood formation or tree growth.
We compared the significant SNPs identified in multi-SNP models and epistasis models and found that of the 24 significant SNP–SNP pairs representing 21 unique SNPs, 81 % of the loci failed to be detected in the multi-SNP models (Table 4 and Table S4). This is consistent with previous studies showing that most of the epistatic interactions have minor main effects and detecting epistasis without main effects (Xu and Jia 2007). Examination of transcript levels in different tissues showed that Pto-miR530a and Pto-KNAT1 expressed in the same tissues with opposite abundances, supporting the interaction of the miRNA and the mRNA (Gamazon et al. 2012), and indicating the epistatic relationship of the two genes. To further determine whether the genotypes of different SNP pairs from the miRNA and mRNA affected the expression pattern, we used RT-qPCR to measure the transcript levels of Pto-KNAT1 across three genotypes of Pto-miR530a-1359964 and Pto-KNAT1-8474222 (Fig. 8). The results showed that plants with the genotype C (C:C or A:C) in Pto-miR530a-1359964 had no significant differences in the transcript levels across three genotypes of Pto-KNAT1-8474222, indicating a masking effect whereby the C allele of Pto-miR530a-1359964 prevents the variation in Pto-KNAT1-8474222 from manifesting its effects (Natarajan et al. 2013a, b). The result that the phenotypes of individuals depend strongly on the specific combination of alleles at two SNPs in Pto-miR530a and Pto-KNAT1 separately indicates that this combination may reflect the functional interaction between the miRNA and the mRNA (Phillips 2008).
Conclusions
In this paper, we used SNP-based association to study the function of Pto-miR530a and Pto-KNAT1 and explore their regulatory interaction. We presented a method combining RT-qPCR-based expression profiling, multi-SNP association under additive and dominant effects, and epistasis analysis to investigate the genetic relationship between this miRNA and its target gene. We first used SNP-based association to identify the interactions between the miRNA and its target; this provides a new strategy to elucidate miRNA–target interactions and their effects on phenotypes. Because miRNAs usually target transcription factors that regulate the expression of dozens of genes, it will be of great importance to elucidate the crucial pathways by studying the miRNA and its upstream transcription factors in future work.
Author contribution
Yang XH and Wang BW collected the data and performed statistical analysis; Zhang DQ contributed in the conception and design; Chen JH helped in the data analysis and interpretation; Yang XH wrote the article; Du QZ and Zhang DQ provided critical revision of the article; Zhang DQ obtained funding and is responsible for the article.
Abbreviations
- 5′-UTR:
-
5′-untranslated region
- DBH:
-
Diameter at breast height
- FL:
-
Fiber length
- FW:
-
Fiber width
- fGWAS:
-
Functional genome-wide association studies
- HC:
-
Holocellulose content
- IG:
-
Information gain
- LC:
-
Lignin content
- LD:
-
Linkage disequilibrium
- MFA:
-
Microfiber angle
- miRNAs:
-
MicroRNAs
- MDR3.0.2:
-
Multifactor dimensionality reduction 3.0.2
- PCN :
-
POPCORONA
- PCA:
-
Principle component analysis
- RT-qPCR:
-
Reverse transcription quantitative PCR
- SNP:
-
Single nucleotide polymorphism
- V:
-
Stem volume
- H:
-
Tree height
References
Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ (2008) Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol 18:758–762
Aukerman MJ, Sakai H (2003) Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 15:2730–2741
Babu M, Arnold R, Bundalovic-Torma C, Gagarinova A, Wong KS, Kumar A, Stewart G, Samanfar B, Aoki H, Wagih O, Vlasblom J, Phanse S, Lad K, Yu A, Graham C, Jin K, Brown E, Golshani A, Kim P, Moreno-Hagelsieb G, Greenblatt J, Houry WA, Parkinson J, Emili A (2014) Quantitative genome-wide genetic interaction screens reveal global epistatic relationships of protein complexes in Escherichia coli. PLoS Genet 2(10):e1004120
Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP (2008) The impact of microRNAs on protein output. Nature 455:64–71
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Bateson W (1909) Mendel’s principles of heredity. Cambridge Univ Press, Cambridge
Beaulieu J, Doerksen T, Boyle B, Clement S, Deslauriers M, Beauseigle S, Blais S, Poulin PL, Lenz P, Caron S, Rigault P, Bicho P, Bousquet J, MacKay J (2011) Association genetics of wood physical traits in the conifer white spruce and relationships with gene expression. Genetics 188:197–214
Brennecke J, Stark A, Russell RB, Cohen SM (2005) Principles of microRNA-target recognition. PLoS Biol 3:e85
Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M et al (2005) A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 353:1793–1801
Chamary HV, Hurst LD (2009) The price of silent mutations. Sci Am 300:46–53
Chen X (2004) A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303:2022–2025
Choi WY, Giraldezb AJ, Schier AF (2007) Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science 318:271–274
Chou HH, Chiu HC, Delaney NF, Segrè D, Marx CJ (2011) Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science 332(6034):1190–1192
Collins RL, Hu T, Wejse C, Sirugo G, Williams SM, Moore JH (2013) Multifactor dimensionality reduction reveals a three-locus epistatic interaction associated with susceptibility to pulmonary tuberculosis. BioData Min 6:4
Donnelly P (2008) Progress and challenges in genome-wide association studies in humans. Nature 465:728–731
Du J, Mansfield SD, Groover AT (2009) The Populus homeobox gene ARBORKNOX2 regulates cell differentiation during secondary growth. Plant J 60:1000–1014
Du J, Miura E, Robischon M, Martinez C, Groover A (2011) The Populus Class III HD ZIP transcription factor POPCORONA affects cell differentiation during secondary growth of woody stems. PLoS ONE 6:e17458
Du Q, Wang B, Wei Z, Zhang D, Li B (2012) Genetic diversity and population structure of Chinese white poplar (Populus tomentosa) revealed by SSR markers. J Hered 103:853–862
Du Q, Xu B, Pan W, Gong C, Wang Q, Tian J, Li B, Zhang D (2013) Allelic variation in a cellulose synthase gene (PtoCesA4) associated with growth and wood properties in Populus tomentosa. G3 3:2069–2084
Du Q, Xu B, Gong C, Yang X, Pan W, Tian J, Li B, Zhang D (2014) Variation in growth, leaf and wood-property traits of Chinese white poplar (Populus tomentosa Carr.), a major industrial tree species in Northern China. Can J Forest Res 44:326–339
Duan R, Pak C, Jin P (2007) Single nucleotide polymorphism associated with mature miR-125a alters the processing of primiRNA. Hum Mol Genet 16:1124–1131
Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP (2005) The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science 310:1817–1821
Gamazon ER, Ziliak D, Im HK, LaCroix B, Park DS, Cox NJ, Huang RS (2012) Genetic architecture of microRNA expression: implications for the transcriptome and complex traits. Am J Hum Genet 90:1046–1063
Groover AT, Mansfield SD, DiFazio SP, Dupper G, Fontana JR, Millar R, Wang Y (2006) The Populus homeobox gene ARBORKNOX1 reveals overlapping mechanisms regulating the shoot apical meristem and the vascular cambium. Plant Mol Biol 61:917–932
Huang ZH (1992) The study on the climatic regionalization of the distributional region of Populus tomentosa. J Beijing Forestry University 14:26–32
Jakulin A, Bratko I (2003) Analyzing attribute dependencies. Lecture Notes in Computer Science, vol 2838. Springer, Berlin Heidelberg, pp 229–240
Jakulin A, Bratko I, Smrke D, Demšar J, Zupan B (2003) Attribute interactions in medical data analysis. Lecture Notes in Computer Science, vol 2780. Springer, Berlin Heidelberg, pp 229–238
Jazdzewski K, Murray EL, Franssila K, Jarzab B, Schoenberg DR, de la Chapelle A (2008) Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci USA 105:7269–7274
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53
Kimchi-Sarfati C, Oh JM, Kim IW, Sauna ZE, Calcagno AM et al (2007) A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315:525–528
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68–D73
Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N (2005) Combinatorial microRNA target predictions. Nature Genet 37:495–500
Lai EC, Tam B, Rubin GM (2005) Pervasive regulation of Drosophila Notch target genes by GY-box-, Brd-box-, and K-box-class microRNAs. Genes Dev 19:1067–1080
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB (2003) Prediction of mammalian microRNA targets. Cell 115:787–798
Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20
Li JH, Das K, Fu G, Li R, Wu RL (2010) The Bayesian Lasso for genome-wide associations studies. Bioinformatics 27:516–523
Liebsch D, Sunaryo W, Holmlund M, Norberg M, Zhang J, Hall HC, Helizon H, Jin X, Helariutta Y, Nilsson O, Polle A, Fischer U (2014) Class I KNOX transcription factors promote differentiation of cambial derivatives into xylem fibers in the Arabidopsis hypocotyl. Development 141:4311–43199
Lim LP, Lau NC, Weinstein EG, Abdelhakim A, Yekta S, Rhoades MW, Burge CB, Bartel DP (2003) The microRNAs of Caenorhabditis elegans. Genes Dev 17:991–1008
Liu L, Zinkgraf M, Petzold HE, Beers EP, Filkov V, Groover A (2015) The Populus ARBORKNOX1 homeodomain transcription factor regulates woody growth through binding to evolutionarily conserved target genes of diverse function. New Phytol 205(2):682–694
Lu SF, Sun YH, Shi R, Clark C, Li LG, Chiang VL (2005) Novel and mechanical stress–responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell 17:2186–2203
Lu SF, Sun YH, Chiang VL (2008) Stress-responsive microRNAs in Populus. Plant J 55:131–151
Lu S, Li Q, Wei H, Chang MJ, Tunlaya-Anukit S, Kim H, Liu J, Song J, Sun YH, Yuan L, Yeh TF, Peszlen I, Ralph J, Sederoff RR, Chiang VL (2013) Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc Natl Acad Sci USA 110:10848–10853
Maher B (2008) Personal genomes: the case of the missing R 2. Nature 456:18–21
Natarajan C, Inoguchi N, Weber RE, Fago A, Moriyama H, Storz JF (2013a) Epistasis among adaptive mutations in deer mouse hemoglobin. Science 340:1324–1327
Natarajan R, Trivedi-Vyas D, Wairkar YP (2013b) Tuberous sclerosis complex regulates Drosophila neuromuscular junction growth via the TORC2/Akt pathway. Human Mol Genet 22:2010–2023
Palatnik JF, Allen E, Wu X, Schommer C, Schwab R, Carrington JC, Weigel D (2003) Control of leaf morphogenesis by microRNAs. Nature 425:257–263
Phillips PC (2008) Epistasis–the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet 9:855–867
Robischon M, Du J, Miura E, Groover A (2011) The Populus class III HD ZIP, popREVOLUTA, influences cambium initiation and patterning of woody stems. Plant Physiol 155:1214–1225
Robnik-Siknja M, Kononenko I (2003) Theoretical and empirical analysis of ReliefF and RReliefF. Mach Learn 53:23–69
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D (2005) Specific effects of microRNAs on the plant transcriptome. Dev Cell 8:517–527
Scofield S, Murray JAH (2006) KNOX gene function in plant stem cell niches. Plant Mol Biol 60:929–946
Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455:58–63
Sexton TR, Henry RJ, Harwood CE, Thomas DS, McManuset LJ, Raymond C, Henson M, Shepherd M (2012) Pectin Methyltransferase genes influence solid wood properties of Eucalyptus pilularis. Plant Physiol 158:531–541
Song YP, Ma KF, Ci D, Zhang ZY, Zhang DQ (2013) Sexual dimorphism floral microRNA profiling and target gene expression in and romonoecious poplar (Populus tomentosa). PLoS ONE 8:e62681
Southam L, Rodriguez-Lopez J, Wilkins JM, Pombo-Suarez M, Snelling S, Gomez-Reino JJ, Chapman K, Gonzalez A, Loughlin J (2007) An SNP in the 5′-UTR of GDF5 is associated with osteoarthritis susceptibility in Europeans and with in vivo differences in allelic expression in articular cartilage. Hum Mol Genet 16:2226–2232
Southerton SG, MacMillan CP, Bell JC, Bhuiyan N, Dowries G, Ravenwood IC, Joyce KR, Williams D, Thumma BR (2010) Association of allelic variation in xylem genes with wood properties in Eucalyptus nitens. Austral Forestry 73:259–264
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet 28:286–289
Thumma BR, Nolan MF, Evans R, Moran GF (2005) Polymorphisms in cinnamoyl CoA reductase (CCR) are associated with variation in microfibril angle in Eucalyptus spp. Genetics 171:1257–1265
Tian JX, Du QZ, Chang MQ, Zhang DQ (2012) Allelic variation in PtGA20Ox associates with growth and wood properties in Populus spp. PLoS ONE 7:e53116
Xu S, Jia Z (2007) Genomewide analysis of epistatic effects for quantitative traits in barley. Genetics 175:1955–1963
Zhang L, Liu Y, Song F, Zheng H, Hu L, Lu H, Liu P, Hao X, Zhang W, Chen K (2011) Functional SNP in the microRNA-367 binding site in the 3′ UTR of the calcium channel ryanodine receptor gene 3 (RYR3) affects breast cancer risk and calcification. Proc Natl Acad Sci USA 108:13653–13658
Zhou L, Bawa R, Holliday JA (2014) Exome resequencing reveals signatures of demographic and adaptive processes across the genome and range of black cottonwood. Mol Ecol 23:2486–2499
Zuckerkandl E, Pauling L (1965) Evolutionary divergence and convergence in proteins. Evolving Genes and Proteins 97:97–166
Acknowledgments
We owe thanks to Prof. Rongling Wu and Dr. Zhong Wang (Center for Statistical Genetics, Pennsylvania State University, Hershey, Pennsylvania) for advice on statistical analysis. This work was supported by the National ‘863’ Plan Project (No. 2013AA102702), the State Key Basic Research Program of China (No. 2012CB114506), and the Project of the National Natural Science Foundation of China (No. 31170622, 30872042).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yang, X., Du, Q., Chen, J. et al. Association mapping in Populus reveals the interaction between Pto-miR530a and its target Pto-KNAT1 . Planta 242, 77–95 (2015). https://doi.org/10.1007/s00425-015-2287-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-015-2287-3