
Abstract
Main conclusion
Identification and characterization of 5,446 mlncRNAs from Salvia miltiorrhiza showed that the majority of identified mlncRNAs were stress responsive, providing a framework for elucidating mlncRNA functions in S. miltiorrhiza.
mRNA-like noncoding RNAs (mlncRNAs) are transcribed by RNA polymerase II and are polyadenylated, capped and spliced. They play important roles in plant development and defense responses. However, there is no information available for mlncRNAs in Salvia miltiorrhiza Bunge, the first Chinese medicinal material entering the international market. To perform a transcriptome-wide identification of S. miltiorrhiza mlncRNAs, we assembled over 8 million RNA-seq reads from GenBank database and 5,624 ESTs from PlantGDB into 44422 unigenes. Using a computational identification pipeline, we identified 5446 S. miltiorrhiza mlncRNA candidates from the assembled unigenes. Of the 5446 mlncRNAs, 2 are primary transcripts of conserved miRNAs, and 2030 can be grouped into 470 families with at least two members in a family. Quantitative real-time PCR analysis of mlncRNAs with at least 900 nt showed that the majority were differentially expressed in roots, stems, leaves and flowers and responsive to methyl jasmonate (MeJA) treatment in S. miltiorrhiza. Analysis of published RNA-seq data showed that a total of 3,044 mlncRNAs were expressed in hairy roots of S. miltiorrhiza and the expression of 1,904 of the 3,044 mlncRNAs was altered by yeast extract and Ag+ treatment. The results indicate that the majority of mlncRNAs are involved in plant response to stress. This study provides a framework for understanding the roles of mlncRNAs in S. miltiorrhiza.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, the next-generation sequencing and transcriptome analysis of eukaryotic cells have provided a large body of evidence for the versatility of RNA molecules. The idea that RNAs not only encode proteins but also function as noncoding RNAs (ncRNAs) is now widely accepted. For instance, more than 90 % of the human genome is likely to be transcribed, but only about 2 % of the transcribed regions encode proteins (Birney et al. 2007). It strongly suggests that there are a large number of RNAs with little or no protein-coding capacity in human cells. In fact, using genomic tiling arrays, whole transcriptome analysis, reverse-transcription PCR (RT-PCR) and computational prediction, ncRNAs acting mainly or exclusively at the RNA level have been identified from various organisms (Bertone et al. 2004; Tran et al. 2009). With the progress of ncRNA identification, it has been shown that the more complex organisms produce the greater proportion of ncRNAs in transcriptomes. Although the function of the majority of ncRNAs is still unknown, current evidences suggest the significance of ncRNAs in plant development and response to environmental stresses (Matsui et al. 2013).
Based on the expression profiles and predicted functions, ncRNAs can be subdivided into housekeeping ncRNAs and regulatory ncRNAs. Housekeeping ncRNAs include tRNAs, rRNAs, snRNAs and snoRNAs. According to the distribution of length, regulatory ncRNAs can be divided into small RNAs (sRNAs) (<200 nt), such as microRNAs (miRNAs) and small-interfering RNAs (siRNAs), and long noncoding RNAs (lncRNAs) (>200 nt), such as mRNA-like noncoding RNAs (mlncRNAs) (Erdmann et al. 2000; Rodriguez et al. 2004; Griffiths-Jones 2007; Rymarquis et al. 2008; Ponting et al. 2009). sRNAs, particularly miRNAs, have been intensively studied. In plants, miRNAs are about 21-nt-long single-stranded small RNAs generated from long primary miRNAs (pri-miRNAs) (Bartel 2004; Voinnet 2009). To produce miRNAs, pri-miRNAs are first transcribed from miRNA loci and fold into internal hairpin structures. Then, Dicer-like 1 (DCL1)-mediated cleavage of pri-miRNAs is carried out. It results in the production of miRNA precursors (pre-miRNAs). Further process of pre-miRNAs by DCL1 and other proteins generates miRNA:miRNA* duplexes bearing 2 nt overhangs at both 3′ ends. The duplexes are finally unwound by a helicase to release the single-stranded mature miRNAs. Plant miRNAs mainly function as posttranscriptional regulators by targeting mRNAs for direct cleavage at a region with perfect or near-perfect sequence complementarities (Bartel 2004). mlncRNAs are a class of lncRNAs transcribed by RNA polymerase II and polyadenylated, capped and spliced as conventional mRNAs (Erdmann et al. 2000; Rodriguez et al. 2004; Griffiths-Jones 2007; Rymarquis et al. 2008). They have all the properties of mRNAs, but lack protein-coding potential (Griffiths-Jones 2007). mlncRNAs may generate from mlncRNA loci located between protein-coding genes. Noncoding alternative transcripts of protein-coding genes, natural antisense transcripts and pri-miRNAs can also be mlncRNAs if they have the properties of mRNAs (Rodriguez et al. 2004; Griffiths-Jones 2007). Compared with miRNA, mlncRNAs are less studied, and owing to their homology to known genes, some mlncRNAs, such as natural antisense transcripts, are often discarded in computational screens (Rymarquis et al. 2008). The majority of currently known plant mlncRNAs were identified from Arabidopsis thaliana (Maclntosh et al. 2001; Ben Amor et al. 2009), Medicago truncatula (Wen et al. 2007), wheat (Xin et al. 2011), Panax ginseng (Wang et al. 2014), Digitalis purpurea (Wu et al. 2012a) and Zea mays (Boerner and McGinnis 2012). A few were obtained from cucumber, soybean and other plants species (Rohrig et al. 2002; Sugiyama et al. 2003; Cho et al. 2005; Ma et al. 2008).
Plant mlncRNAs appear to play significant roles in cell differentiation, plant response to biotic and abiotic stresses and nodulation. For instance, cucumber CR20, one of the first transcripts identified as plant mlncRNAs, was repressed by cytokinins (Teramoto et al. 1996). GUT15 (gene with unstable transcript 15), known as an unstable transcripts in tobacco cell cultures, was hormone-regulated (Taylor and Green 1995). CsM10 was found to be highly expressed in the apexes of one and four leaf cucumber seedlings grown under short day (i.e. male-inducing) conditions than under long day (i.e. female-inducing) conditions in Cucumis sativus (Cho et al. 2005). CCLS96.1 was expressed in both genders in leaves and flowers in Silene latifolia (Sugiyama et al. 2003). Zm401 played important roles in pollen development in Zea mays (Ma et al. 2008). TPSI1/Mt4 responded to phosphate starvation in tomato (Liu et al. 1997) and Medicago (Burleigh and Harrison 1997). Moreover, some mlncRNAs functioned as endogenous target mimics (eTMs) to inhibit the activity of miRNAs in plants. So far, a total of 36 Arabidopsis mlncRNAs have been identified to be potential eTMs for 11 miRNAs and 189 rice mlncRNAs to be eTMs for 19 miRNAs. Overexpression of IPS1, one of Arabidopsis eTMs, resulted in the accumulation of miR399-targeted PHO2 transcripts in Arabidopsis (Wu et al. 2013).
Salvia miltiorrhiza Bunge is a well-known Salvia species in the Labiatae family with great economic and medicinal value. It has been widely used in the treatment of cardiovascular, hyperlipidemia, cerebrovascular and acute ischemic stroke diseases, and is the first Chinese medicinal material entering the international market (Li et al. 2009b). The active pharmaceutical ingredients of S. miltiorrhiza mainly include lipophilic tanshinones, such as tanshinone I and tanshinone IIA, and hydrophilic phenolic acids, such as rosmarinic and salvianolic acids (Wu et al. 2012b). It has been shown that methyl jasmonate (MeJA), an effective elicitor, could enhance the accumulation of tanshinones and phenolic acids through inducing the expression of various tanshinone biosynthesis- and phenolic acid biosynthesis-related genes (Luo et al. 2014). Moreover, the biosynthesis of tanshinones in S. miltiorrhiza hairy root cultures could also be enhanced by biotic (yeast extract) and abiotic (Ag+) treatments (Gao et al. 2014).
Salvia miltiorrhiza is an emerging model system for medicinal plant biology because of its relatively small genome size (about 600 Mb), short life cycle, undemanding growth requirements and significant medicinal value (Song et al. 2013). S. miltiorrhiza genes involved in secondary metabolism (Ma et al. 2012; Li and Lu 2014; Zhang et al. 2014a, b), stress response (Zhao et al. 2014) and miRNA pathway (Shao and Lu 2013, 2014) have been intensively studied recently. However, there is no information available for S. miltiorrhiza mlncRNAs. With the aim to identify mlncRNAs in S. miltiorrhiza, a large number of expressed sequences collected in GenBank database and PlantGDB were assembled. A total of 44,422 unigenes were obtained, from which 5,446 mlncRNAs were identified. It includes two pri-miRNAs. Various important characteristics of the identified mlncRNAs were analyzed using computational and experimental approaches. The results suggest that mlncRNAs are functionally meaningful in S. miltiorrhiza.
Materials and methods
Plant materials and stress treatment
Salvia miltiorrhiza Bunge (line 993, plants kindly provided by Prof. Xian’en Li) was grown in a field nursery at the Institute of Medicinal Plant Development, Beijing, China (Ma et al. 2012). Flowers, healthy leaves, young stems and roots with diameters about 0.5 cm were collected from 2-year-old plants. Samples from three plants were pooled and stored in liquid nitrogen until use. Plantlet cultivation and methyl jasmonate (MeJA) treatment were performed as described previously (Ma et al. 2012). Briefly, stem segments of field-grown S. miltiorrhiza (line 993) were used as explants for generation of shoots on MS agar medium (Murashige and Skoog 1962). Small shoots were excised from the explants and then rooted on 6.7-V agar medium (Chen et al. 1997). About 6-week-old plantlets with regenerated roots were transferred to 6.7-V liquid medium and cultivated for 2 days. MeJA in carrier solution containing 0.1 % Tween-20 and 5 % ethanol was added to the medium to obtain a final concentration of 200 µM. Plantlets were treated for 12, 24, 36 and 48 h and then leaves with similar sizes were collected. Plantlets treated with carrier solution containing 0.1 % Tween-20 and 5 % ethanol were used as controls. Three biological replicates were carried out for each experiment.
EST assembly, annotation and classification
RNA-seq reads were downloaded from GenBank database under the accession numbers SRA020132 (Hua et al. 2011) and SRA012102 (Li et al. 2010). S. miltiorrhiza EST sequences were downloaded from PlantGDB (http://www.plantgdb.org/). High-quantity RNA-seq reads and ESTs were assembled into unigenes using Trinity software. The sequences of 5 SmRDRs (Shao and Lu 2014), 10 SmAGOs (Shao and Lu 2013) and 110 SmMYBs (Li and Lu 2014) were downloaded from GenBank under the accession numbers KF872203–KF872207, KF153679–KF153688, and KF059355–KF059464, respectively. Sequence comparison of the assembled unigenes with SmRDRs, SmAGOs and SmMYBs was analyzed using BLASTN (Altschul et al. 1997). An e value cutoff of 10−5 was applied to the homolog recognition. Unigenes were annotated by sequence similarity searched against the NCBI non-redundant protein (Nr) database, Swiss-Prot protein database, Gene Ontology (GO) database, KEGG database and COG database using the Basic Local Alignment Search Tool (BLAST) (Altschul et al. 1997). An e value cutoff of 10−5 was applied to the homolog recognition. Based on the annotation results from BLAST search against the Nr database, Gene Ontology (GO) terms were assigned by Blast2Go software (Conesa et al. 2005) and classified functionally by WEGO software (Ye et al. 2006).
Computational identification of mlncRNAs
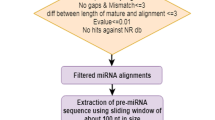
The computational method for mlncRNA identification is shown in Fig. 1. The assembled unigenes without Nr annotation were first checked for simple repeat transcripts and then open reading frames (ORFs) were predicted using ESTScan2 software with the cutoff of 100 amino acids (aa) (Iseli et al. 1999; Maclntosh et al. 2001). Computational prediction was performed on linux system using default parameters. To distinguish mlncRNA candidates from housekeeping ncRNAs, unigenes with the longest ORF less than 100 aa were analyzed against Rfam 11.0 (August 2012) using BLASTN (Altschul et al. 1997; Gardner et al. 2011). The conservation of mlncRNAs was analyzed by searching S. miltiorrhiza mlncRNA candidates against the NONCODE database and PLncDB database using BLASTN (Altschul et al. 1997; He et al. 2008; Jin et al. 2013). A word size 7 and an e value cutoff of 10−5 were applied to the homolog recognition.

Workflow of the filtering process used to identify mlncRNA candidates in S. miltiorrhiza. The number of unigenes is shown in parentheses. ORF open reading frame, aa amino acid
Identification of primary transcripts of conserved miRNAs
Plant miRNA sequences were downloaded from miRBase (Released 19, http://www.mirbase.org/). mlncRNA candidates were searched against plant miRNA sequences with no more than three mismatches using the BLASTN program. A word size of 4 was used as described. Secondary structures of mlncRNA candidates were predicated on the mfold web server (http://www.bioinfo.rpi.edu/applications/mfold) and then checked manually (Zuker 2003). The criteria applied include (1) a single-stranded, hairpin precursor with the free energy value less than −30, (2) four or fewer mismatched miRNA bases, and (3) less than two asymmetric bulges within the miRNA/miRNA* duplex (Meyers et al. 2008). The targets of miRNAs were predicted on the psRNATarget Server using the default parameters with the maximum expectation to be 2.5 (Dai et al. 2011).
Quantitative real-time reverse-transcription PCR (qRT-PCR) analysis
Total RNA was extracted from plant tissues using the plant total RNA extraction kit (Aidlab, Beijing, China) and digested with RNase-free DNase I (TaKaRa Bio, Otsu, Japan) to remove genomic DNA contamination. RNA integrity was analyzed on a 1 % agarose gel and RNA quantity was determined using a NanoDrop 2000C Spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Reverse transcription was performed on 1.0 µg total RNA for each example by 200 U SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) in 20 µl volume. The resulting cDNA was diluted to 200 µl with sterile water. Quantitative real-time PCR was carried out in triplicate reactions in the CFX96™ real-time PCR detection system (Bio-Rad, Hercules, CA, USA). Gene-specific primers were designed using the primer designing tools of IDT dna (http://www.idtdna.com/scitools/Applications/RealTimePCR/). Primer sequences are listed in Supplementary Table S1. SmUBQ10 was used as a reference as described previously (Ma et al. 2012; Shao and Lu 2013). PCR was carried out in 20 µl volume containing 2 µl diluted cDNA, 0.5 µM forward primer, 0.5 µM reverse primer, and 1× SYBR Premix Ex Taq II (Tli RNaseH Plus) (TaKaRa Bio) using the following conditions: 95 °C for 30 s, 40 cycles of 95 °C for 5 s, 60 °C for 18 s and 72 °C for 15 s. Amplification specificity was assessed using the dissociation curve. Relative abundance of transcripts was analyzed using the comparative Cq method (Livak and Schmittgen 2001). The arithmetic formula 2−ΔΔCq was used to achieve results for relative quantification. Cq represents the threshold cycle. Gene expression data from three biological replicates were standardized as described (Gardner et al. 2011). ANOVA (analysis of variance) was calculated using SPSS (Version 19.0). P < 0.05 was considered statistically significant.
Analysis of mlncRNA expression in response to yeast extract and Ag+ treatment
RNA-seq data obtained from S. miltiorrhiza hairy roots treated with yeast extract (100 µg/ml) and Ag+ (30 µM) were downloaded from GenBank under the accession number SRR924662 (Gao et al. 2014). RNA-seq reads from non-treated (0 hpi) and treated for 12 h (12 hpi), 24 h (24 hpi) and 36 h (36 hpi) were mapped to mlncRNAs using the SOAP2 software (Li et al. 2009a) with the parameter v cutoff of 3 and parameter r cutoff of 2. The expression level of mlncRNAs at different time-points was calculated by RPKM (RNA-seq reads mapped to an mlncRNA per total million reads from a treatment per kilobases of the mlncRNA length). mlncRNAs with the log-2-transformed RPKM value greater than 2 were selected for differential expression analysis. RNA-seq reads of an mlncRNA in yeast extract and Ag+-treated tissues were compared with the reads in non-treated tissues using Fisher’s exact test. mlncRNAs with the Fisher’s exact test P value of less than 0.05 were considered as differentially expressed.
Results
Assembly and functional annotation of the S. miltiorrhiza transcriptome
To explore the transcriptome of S. miltiorrhiza, we collected over 8 million RNA-seq sequences from GenBank under the accession numbers SRA020132 (Hua et al. 2011) and SRA012102 (Li et al. 2010) and 5624 ESTs from PlantGDB (http://www.plantgdb.org). After pre-processing and de novo assembly using Trinity software, a total of 44,422 unigenes (≥200 bp) were obtained. N50 length of the assembled unigenes was 550 bp, and the average length was 501 bp. The distribution of unigene sizes showed that most of the assembled unigenes ranged from 200 to 600 bp (Supplementary Fig. S1). Sequence comparison revealed high identities between the assembled unigenes and recently reported cDNAs, including 5 SmRDRs (Shao and Lu 2014), 10 SmAGOs (Shao and Lu 2013) and 110 SmMYBs (Li and Lu 2014) (Supplementary Table S2). It suggests that the RNA-seq data have been successfully assembled.
Functional annotation of unigenes was carried out by BLAST analysis (Altschul et al. 1997) against various databases, including the Nr database, Swiss-Prot protein databases, GO database, KEGG database and COG database. An e value cutoff of 10−5 was applied to the homolog recognition. It resulted in the annotation of 34,615 unigenes, which accounted for 77.92 % of total unigenes (Table 1). The remainders which did not show significant similarity to any sequences in the protein databases could be novel protein-coding transcripts, UTR regions of known protein-coding genes, partial ORFs and UTR regions of protein-coding genes, or ncRNAs.
GO term, an international standard classified system in terms of its associated biological process, cellular component and molecular function, was used in functional assignment and classification of the assembled unigenes. Among the 27,156 unigenes annotated by GO, 21,734 were assigned to biological process, 23,293 to cellular component, and 20,699 to molecular function. In some cases, multiple terms were assigned to a unigene. GO assignments of 55 subcategories of three categories are shown in Supplementary Fig. S2. Among the biological process categories, the majority are related to cellular process (15.38 %) and metabolic process (14.58 %). Cell (24.66 %) is the greatest subcategory in the cellular component. Within the molecular function category, catalytic activity (41.88 %) was assigned the most number of unigenes. The results provide abundant information for the function of S. miltiorrhiza genes.
Computational prediction of mlncRNAs
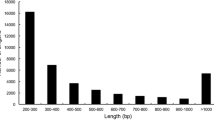
To identify S. miltiorrhiza mlncRNAs, the 11464 unigenes without Nr annotation were first mapped to the current S. miltiorrhiza genome assembly, which covers about 92 % of the entire genome and 96 % of the protein-coding genes (Song et al. 2013). Eighty-three unigenes that could not be mapped were discarded. To remove the unigenes probably generated from UTR regions of known protein-coding genes or including partial ORFs and UTRs of protein-coding genes, S. miltiorrhiza genomic DNA sequences mapped with unigenes were extended 1,000 bp at both ends of unigenes and then annotated by BLAST analysis (Altschul et al. 1997) against the Nr database. It resulted in the identification of 5,468 unigenes probably produced from protein-coding genes. The remaining 5,913 unigenes were then subjected to detailed characterization using the pipeline summarized in Fig. 1. The pipeline we used is similar to those reported previously for systematic identification of mlncRNAs from EST databases of A. thaliana (Maclntosh et al. 2001; Ben Amor et al. 2009), M. truncatula (Wen et al. 2007), wheat (Xin et al. 2011), P. ginseng (Wang et al. 2014), D. purpurea (Wu et al. 2012a) and Z. mays (Boerner and McGinnis 2012). The pipeline recognized 3 unigenes encoding simple repeats and 452 putative novel protein genes. It also identified 15 housekeeping ncRNAs, including a tRNA precursor, 2 rRNA precursors and 12 snoRNA precursors (Table 2). The identification of small number of housekeeping ncRNAs suggests that the majority of unigenes obtained in this study are derived from polyadenylated transcripts. The remaining 5,446 unigenes were considered as mlncRNA candidates (Supplementary Table S3). Size distribution analysis showed that the majority of mlncRNAs were between 200 bp and 600 bp in length (Fig. 2). Although we cannot rule out the possibility that some of the mlncRNA candidates, particularly the shorter ones, were fragments of protein-coding transcripts, the longer mlncRNA candidates, such as those with length over 900 bp (Table 3), are very likely to be authentic (Wu et al. 2012a).

Size distribution of the identified mlncRNA candidates in S. miltiorrhiza
Identification of two pri-miRNAs
miRNAs, a class of small endogenous noncoding RNAs playing crucial regulatory roles in plant development and stress responses, originate from long primary transcripts (Bartel 2004; Lu et al. 2008). To determine whether there were some mlncRNAs to be primary transcripts of miRNA, we carried out computational search of mlncRNAs against plant miRNA sequences in miRBase (Released 19, http://www.mirbase.org/) using the BLASTN program (Altschul et al. 1997) and analyzed the secondary structures of mlncRNAs with regions no more than three mismatches to known plant miRNAs using mfold (http://www.bioinfo.rpi.edu/applications/mfold) (Zuker 2003). Manually checking the secondary structures with the criteria for plant miRNA annotation (Meyers et al. 2008) resulted in the identification of two pri-miRNAs (Unigene16661_All and Unigene5483_All), all of which had the potential to generate members of the deeply conserved MIR156 family (Fig. 3). It is possible that some other mlncRNAs are primary transcripts of novel miRNAs, which need to be further examined.

Predicted hairpin structures of S. miltiorrhiza miRNA precursors. Mature miRNA sequences are indicated in red. The red, green and blue vertical lines indicate G:C, G:U and A:U pairing, respectively
In plants, the regulatory role of miRNAs is mainly through targeting transcripts for direct cleavage at a region with perfect or near-perfect complementaries between miRNAs and their targets (Bartel 2004). It allows an effective prediction of miRNA targets through computation. Using psRNATarget with the default parameters with the maximum expectation to be 2.5 (Dai et al. 2011), 2 (Unigene9660_All and Unigene16386_All) of the 44,422 unigenes were predicted to be targets of S. miltiorrhiza miR156a and miR156b. All of them encode SQUAMOSA promoter binding protein-likes. It is consistent with previous results showing the cleavage of miR156 on S. miltiorrhiza SPL transcripts (Zhang et al. 2014a). These results suggest that miR156 plays crucial regulatory roles in the development of S. miltiorrhiza and indicate that some mlncRNAs are important regulators in S. miltiorrhiza.
Characterization of S. miltiorrhiza mlncRNA sequences
Members of a gene family can be derived from the same ancestors and exhibit similarity on structures and functions (Wu et al. 2012a; Zhang et al. 2014a). To obtain the first clue to the function of mlncRNAs, we classified the identified 5,444 S. miltiorrhiza mlnRNAs (two pri-miRNAs excluded) based on the sequence homology. The results showed that 2,030 of the 5,444 mlncRNAs could be grouped into 470 families with at least two members in a family (Supplementary Table S4). The number of members in a family is varied from 2 to 530. No homologs were found for the other 3,414 mlncRNAs. It suggests that the majority of mlncRNAs exist as a single copy in the S. miltiorrhiza genome and indicates that some mlncRNAs may be recently evolved.
Although many S. miltiorrhiza mlncRNAs show sequence similarity and can be grouped into families, the majority of mlncRNAs exhibit low conservation among organisms, such as A. thaliana (Maclntosh et al. 2001), M. truncatula (Wen et al. 2007), wheat (Xin et al. 2011), D. purpurea (Wu et al. 2012a), and Z. mays (Boerner and McGinnis 2012). To determine the conservation of S. miltiorrhiza mlncRNAs, we searched the NONCODE database of known ncRNAs and PLncDB database of plant lncRNAs for homologs of the 5444 mlncRNA candidates (pri-miRNAs excluded) using BLASTN (Altschul et al. 1997; He et al. 2008; Jin et al. 2013). A total of 32 mlncRNAs were found to be conserved (Table 4). Among them, three, including CL3766.Contig2_ALL, CL4282.Contig1_All and CL4282.Contig1_All, are homologues of hormone-regulated unstable transcripts GUT15 (Taylor and Green 1995). Two, including Unigene14038_All and Unigene7516_All, are homologues of the Pi starvation-responsive TPSI1 (Liu et al. 1997). The other 27 conserved mlncRNAs are homologues of other functionally unknown noncoding RNAs.
Tissue specificity of mlncRNA expression
Using the qRT-PCR, expression patterns of 18 mlncRNAs with size over 900 bp were analyzed in flowers, leaves, stems and roots of 2-year-old field nursery-grown S. miltiorrhiza plants. Of the 18 mlncRNAs, mlncR12 were undetected, suggesting that it could be not expressed or expressed at a low level in the tissues analyzed. The other 17 were detected in at least a tissue and showed tissue-specific expression (Fig. 4). mlncR1, mlncR2 and mlncR5 were expressed mainly in flowers. mlncR4, mlncR6, mlncR7, mlncR11, mlncR14, mlncR15 and mlncR16 were more leaf-specific. mlncR8, mlncR10 and mlncR17 exhibited the highest expression in roots. mlncR9 was expressed predominantly in leaves and stems. mlncR13 and mlncR18 showed higher expression in stems, leaves and roots, less in flowers. The results suggest that the expression of mlncRNAs is developmentally regulated in S. miltiorrhiza.

Expression of mlncRNAs in roots (R), stem (S), leaves (L) and flowers (F) of 2-year-old S. miltiorrhiza plants. Expression levels were quantified by qPCR. Fold changes of mlncRNA levels are shown. The level of transcripts in flowers was arbitrarily set to 1 and the level in other tissues was given relative to this. ANOVA was calculated using SPSS (Version 19.0, IBM, USA). P < 0.05 was considered statistically significant
Transcriptome-wide analysis of mlncRNA expression in response to yeast extract and Ag+ treatment
It has been shown that mlncRNAs are involved in plant response to various stresses, such as cold and dehydration (Wu et al. 2012a). To determine whether the identified S. miltiorrhiza mlncRNAs are stress responsive, we carried out a transcriptome-wide analysis of mlncRNA expression in response to yeast extract and Ag+ treatment. RNA-seq data of S. miltiorrhiza hairy roots treated with or without yeast extract (100 µg/ml) and Ag+ (30 µM) were obtained from GenBank (accession number SRR924662) (Gao et al. 2014) and then mapped to the identified mlncRNAs using SOAP2 (Li et al. 2009a). About 2.68–3.16 % of RNA-seq reads from non-treated (0 hpi) and treated for 12 h (12 hpi), 24 h (24 hpi) and 36 h (36 hpi) were perfectly mapped to mlncRNAs (Table 5). Overall, a total of 3,044 (55.69 %) mlncRNAs were found to be expressed in S. miltiorrhiza hairy roots treated with or without yeast extract (100 µg/ml) and Ag+ (30 µM) (Table 5). The log-2-transformed RPKM value of most expressed mlncRNAs is between 4 and 12 (Fig. 5). Compared with the level in non-treated (0 hpi) control, 1,904 of the 3,044 mlncRNAs expressed in hairy roots were differentially expressed (Fig. 6; Supplementary Table S5). It included 1,244 differentially expressed only at 12 hpi, 43 only at 24 hpi, 28 only at 36 hpi, 216 at 12 hpi and 24 hpi, 141 at 12 hpi and 36 hpi, 32 at 24 hpi and 36 hpi, and 200 at all three time-points. Among the 1,801 mlncRNAs differentially expressed at 12 hpi, 893 were up-regulated, while the other 908 were down-regulated (Table 6). Of the 491 mlncRNAs differentially expressed at 24 hpi, 151 were up-regulated and 340 were down-regulated. Among the 401 mlncRNAs differentially expressed at 36 hpi, the number of up- and down-regulated mlncRNAs was 163 and 238, respectively (Table 6). These results suggest that the majority of mlncRNAs are involved in plant response to stress.

Proportion of expressed mlncRNAs with different log-2-transformed RPKM values. mlncRNAs expressed in S. miltiorrhiza hairy roots treated with yeast extract and Ag+ for 0 h (0 hpi), 12 h (12 hpi), 24 h (24 hpi) and 36 h (36 hpi) are shown

The number of mlncRNAs responsive to yeast extract and Ag+ treatment in S. miltiorrhiza hairy roots. The tissues were treated for 12 h (12 hpi), 24 h (24 hpi) and 36 h (36 hpi). mlncRNAs with the log-2-transformed RPKM value greater than 2 and the Fisher’s exact test P value of less than 0.05 were considered as differentially expressed. Fisher’s exact test was performed on RNA-seq reads of an mlncRNA from yeast extract and Ag+-treated tissues and its reads from non-treated tissues
The response of mlncRNAs to MeJA treatment in S. miltiorrhiza
MeJA is a significant regulator in plant response to biotic and abiotic stresses. It is also an effective elicitor for the production of tanshinones and phenolic acids in S. miltiorrhiza (Gao et al. 2014; Luo et al. 2014). To further analyze the role of mlncRNAs in defense response in S. miltiorrhiza, expression patterns of 17 mlncRNAs with length over 900 bp were analyzed in leaves of plantlets treated with MeJA for 12, 24, 36 and 48 h using the quantitative real-time PCR method. Plantlets treated with carrier solution were used as controls and three independent biological replicates were performed. As shown in Fig. 7, the expression of 15 of the 17 mlncRNAs analyzed was significantly altered in leaves of S. miltiorrhiza plantlets at at least a time-point of MeJA treatment. These 15 mlncRNAs are considered as MeJA-responsive mlncRNAs, of which 12 were significantly down-regulated at different time-points of treatment, such as mlncR7, mlncR8, mlncR9, mlncR14, mlncR16 and mlncR17 at the time-point of 24-h treatment; mlncR1 and mlncR4 at the time-point of 36-h treatment; mlncR5, mlncR10, mlncR11 and mlncR13 at the time-points of 24- and 36-h treatments; mlncR6 at the time-points of 12-, 36- and 48-h treatments; and mlncR2 at all four points of time. It suggests that the majority of mlncRNAs were down-regulated after MeJA treatment. Interestingly, the expression of mlncR18 fluctuated from time-point to time-point. It was significantly down-regulated at the time-point of 24-h treatment, while up-regulated at the time-point of 36-h treatment. These results suggest the significance of mlncRNAs in response to MeJA treatment in S. miltiorrhiza.

Response of mlncRNAs to MeJA treatment. Expression of 17 mlncRNAs with length over 900 bp in the leaves of S. miltiorrhiza plantlets treated with MeJA for 12, 24, 36 and 48 h was shown. The level of transcripts in leaves treated with carrier solution (CK) was arbitrarily set to 1 and the level in tissues treated with MeJA was given relative to this. Fold changes of mlncRNA levels were shown. ANOVA was calculated using SPSS (Version 19.0, IBM, USA). P < 0.05 (*) and P < 0.01 (**) were considered statistically significant
Discussion
Salvia miltiorrhiza Bunge, known as Danshen in Chinese, is an important traditional Chinese medicine and is also an emerging model system for medicinal plant biology (Song et al. 2013). However, there is no information available for mlncRNAs in S. miltiorrhiza. In this study, we identified a total of 5,446 mlncRNA. It is the first attempt to identify mlncRNAs in S. miltiorrhiza. Although it is possible that some of the identified mlncRNAs are false positive, the longer ones are more likely to be authentic. The results provide useful information for further analyzing the function of mlncRNAs in S. miltiorrhiza.
Among the 5,446 mlncRNA candidates, 2,030 can be grouped into families with at least two members in a family (Supplementary Table S4). The number of three biggest mlncRNA families is 530, 62, and 39, respectively. The existence of super large mlncRNA families was also found in P. ginseng (Wang et al. 2014). It indicates the significance and complexity of mlncRNAs in plants. BLAST analysis of the conservation of mlncRNAs against the NONCODE database and PLncDB database showed that the vast majority of mlncRNAs (99.4 %) could be species specific. Similar results were observed for mlncRNAs in other plant species, such as A. thaliana (Maclntosh et al. 2001), M. truncatula (Wen et al. 2007), wheat (Xin et al. 2011), P. ginseng (Wang et al. 2014), D. purpurea (Wu et al. 2012a) and Z. mays (Boerner and McGinnis 2012). It suggests low conservation degree of plant mlncRNAs from different species, although many mlncRNAs from a species show sequence similarity and can be grouped into families.
Although the function of most plant mlncRNAs is still unknown, current results suggest that mlncRNAs may play regulatory roles through interacting with RNA, DNA, and protein-coding genes (Burleigh and Harrison 1997; Ben Amor et al. 2009; Ponting et al. 2009; Kim et al. 2010) or function as small RNA precursors (Wu et al. 2012a; Wang et al. 2014). For example, Arabidopsis IPS1 and M. truncatula Mt4 fine-tune the expression of miR399/PHO2 in plants under phosphate starvation (Burleigh and Harrison 1997; Liu et al. 1997). D. purpurea mlncR8 and mlncR31 contain regions sharing high identities with genes encoding 4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase (HDS) and solanesyl diphosphate synthase (SPS), respectively (Wu et al. 2012a). P. ginseng MAR generates a large amount of small RNAs to regulate targets involved in diverse metabolic pathways (Wang et al. 2014). Moreover, among the 3,688 P. ginseng mlncRNAs, 11 produced miRNAs (Wang et al. 2014). Among the 2,660 D. purpurea mlncRNA candidates, 13 are primary transcripts of conserved miRNAs (Wu et al. 2012a). In this study, two primary transcripts of miR156 were identified from the 5446 S. miltiorrhiza mlncRNA candidates. miR156 regulates S. miltiorrhiza development through direct cleavage of SPL transcripts (Zhang et al. 2014a). The results suggest that some mlncRNAs are important regulators in S. miltiorrhiza. Based on the results from other plant species, such as P. ginseng (Wang et al. 2014), it is very likely that some other S. miltiorrhiza mlncRNAs are primary transcripts of siRNAs or novel miRNAs. These mlncRNAs remain to be identified.
Expression patterns of S. miltiorrhiza mlncRNAs analyzed exhibit apparent tissue specificity. It is consistent with the results from D. purpurea mlncRNAs (Wu et al. 2012a), suggesting that mlncRNAs are developmentally regulated and involved in plant growth and development. Transcriptome-wide analysis of mlncRNA expression showed that the majority of mlncRNAs expressed in hairy roots of S. miltiorrhiza were responsive to yeast extract and Ag+ treatment. Quantitative RT-PCR analysis revealed that the expression levels of 15 of 17 mlncRNAs analyzed were significantly altered in leaves of S. miltiorrhiza plantlets treated with MeJA treatment. Similarly, among the 27 mlncRNAs expressed in D. purpurea plantlets, 24 and 27 are responsive to cold and dehydration, respectively (Wu et al. 2012a). The results suggest the sensitivity of mlncRNA expression in plants growing under the stressful environments and indicate the significance of mlncRNAs in plant response to stress. It has been shown that yeast extract, Ag+ and MeJA are effective elicitors for the production of bioactive compounds in S. miltiorrhiza (Gao et al. 2014). The response of many mlncRNAs in the treatment of these elicitors indicates that some mlncRNAs are probably associated with the biosynthesis of bioactive compounds in S. miltiorrhiza. Further characterization of the identified mlncRNAs may help to test this hypothesis.
Author contribution
SL conceived and designed research. DL, FS and SL analyzed the data. FS performed MeJA treatment. DL conducted experiments. DL contributed reagents, materials and analytical tools. SL and DL wrote the manuscript. All authors read and approved the manuscript.
Abbreviations
- ncRNA:
-
Noncoding RNA
- qRT-PCR:
-
Quantitative reverse-transcription PCR
- sRNA:
-
Small RNA
- miRNA:
-
MicroRNA
- siRNA:
-
Small-interfering RNA
- lncRNA:
-
Long noncoding RNA
- mlncRNA:
-
mRNA-like noncoding RNA
- pri-miRNA:
-
Primary miRNA
- pre-miRNA:
-
miRNA precursor
- DCL1:
-
Dicer-like 1
- MeJA:
-
Methyl jasmonate
- ORF:
-
Open reading frame
- FDA:
-
Food and Drug Administration
- HDS:
-
4-Hydroxy-3-methylbut-2-en-1-yl diphosphate synthase
- SPS:
-
Solanesyl diphosphate synthase
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Ben Amor B, Wirth S, Merchan F, Laporte P, d’Aubenton-Carafa Y, Hirsch J, Maizel A, Mallory A, Lucas A, Deragon JM, Vaucheret H, Thermes C, Crespi M (2009) Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res 19:57–69
Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, Weissman S, Gerstein M, Snyder M (2004) Global identification of human transcribed sequences with genome tiling arrays. Science 306:2242–2246
Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR et al (2007) Identification and analysis of functional elements in 1 % of the human genome by the ENCODE pilot project. Nature 447:799–816
Boerner S, McGinnis KM (2012) Computational identification and functional predictions of long noncoding RNA in Zea mays. PLoS ONE 7:e43047
Burleigh SH, Harrison MJ (1997) A novel gene whose expression in Medicago truncatula roots is suppressed in response to colonization by vesicular–arbuscular mycorrhizal (VAM) fungi and to phosphate nutrition. Plant Mol Biol 34:199–208
Chen H, Yuan JP, Chen F, Zhang YL, Song JY (1997) Tanshinone production in Ti-transformed Salvia miltiorrhiza cell suspension cultures. J Biotechnol 58:147–156
Cho J, Koo DH, Nam YH, Han CK, Lim HT, Bang JW, Hur Y (2005) Isolation and characterization of cDNA clones expressed under male sex expression conditions in a monoecious cucumber plant (Cucumis sativus L. cv. Winter Long). Euphytica 146:271–281
Conesa A, Gotz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis infunctional genomics research. Bioinformatics 21:3674–3676
Dai X, Zhuang Z, Zhao PX (2011) Computational analysis of miRNA targets in plants: current status and challenges. Brief Bioinform 12:115–121
Erdmann VA, Szymanski M, Hochberg A, Groot N, Barciszewski J (2000) Non-coding, mRNA-like RNAs database Y2K. Nucleic Acids Res 28:197–200
Gao W, Sun HX, Xiao H, Cui G, Hillwig M, Jackson A, Wang X, Shen Y, Zhao N, Zhang L, Wang XJ, Peters RJ, Huang L (2014) Combining metabolomics and transcriptomics to characterize tanshinone biosynthesis in Salvia miltiorrhiza. BMC Genom 15:73
Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, Finn RD, Nawrocki EP, Kolbe DL, Eddy SR, Bateman A (2011) Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res 39:D141–D145
Griffiths-Jones S (2007) Annotating noncoding RNA genes. Annu Rev Genomics Hum Genet 8:279–298
He S, Liu C, Skogerbø G, Zhao H, Wang J, Liu T, Bai B, Zhao Y, Chen R (2008) NONCODE v2.0: decoding the non-coding. Nucleic Acids Res 36:D170–D172
Hua W, Zhang Y, Song J, Zhao L, Wang Z (2011) De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 98:272–279
Iseli C, Jongeneel CV, Bucher P (1999) ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol, 138–148
Jin J, Liu J, Wang H, Wong L, Chua NH (2013) PLncDB: plant long non-coding RNA database. Bioinformatics 29:1068–1071
Kim DH, Zografos BR, Sung S (2010) Mechanisms underlying vernalization mediated VIN3 induction in Arabidopsis. Plant Signal Behav 5:1457–1459
Li C, Lu S (2014) Genome-wide characterization and comparative analysis of R2R3-MYB transcription factors shows the complexity of MYB-associated regulatory networks in Salvia miltiorrhiza. BMC Genom 15:277
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J (2009a) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967
Li YG, Song L, Liu M, Hu ZB, Wang ZT (2009b) Advancement in analysis of Salvia miltiorrhizae Radix et Rhizoma (Danshen). J Chromatogr A 1216:1941–1953
Li Y, Sun C, Luo HM, Li XW, Niu YY, Chen SL (2010) Transcriptome characterization for Salvia miltiorrhiza using 454 GS FLX. Yao Xue Xue Bao 45:524–529
Liu C, Muchhal US, Raghothama KG (1997) Differential expression of TPSI1, a phosphate starvation-induced gene in tomato. Plant Mol Biol 33:867–874
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2 (−Delta Delta C(T)) method. Methods 25:402–408
Lu S, Sun YH, Chiang VL (2008) Stress-responsive microRNAs in Populus. Plant J 55:131–151
Luo H, Zhu Y, Song J, Xu L, Sun C, Zhang X, Xu Y, He L, Sun W, Xu H, Wang B, Li X, Li C, Liu J, Chen S (2014) Transcriptional data mining of Salvia miltiorrhiza in response to methyl jasmonate to examine the mechanism of bioactive compound biosynthesis and regulation. Physiol Plant 152:241–255
Ma J, Yan B, Qu Y, Qin F, Yang Y, Hao X, Yu J, Zhao Q, Zhu D, Ao G (2008) Zm401, a short-open reading-frame mRNA or noncoding RNA, is essential for tapetum and microspore development and can regulate the floret formation in maize. J Cell Biochem 105:136–146
Ma Y, Yuan L, Wu B, Li X, Chen S, Lu S (2012) Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza. J Exp Bot 63:2809–2823
Maclntosh GC, Wilkerson C, Green PJ (2001) Identification and analysis of Arabidopsis expressed sequence tags characteristic of non-coding RNAs. Plant Physiol 127:765–776
Matsui A, Nguyen AH, Nakaminami K, Seki M (2013) Arabidopsis non-coding RNA regulation in abiotic stress responses. Int J Mol Sci 14:22642–22654
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao XF, Carrington JC, Chen XM, Green PJ, Griffiths-Jones S, Jacobsen SE, Martienssen RA, Mallory AC, Poethig S, Qi YJ, Vaucheret H, Voinnet O, Watanabe Y, Weigel D, Zhu JK (2008) Criteria for annotation of plant microRNAs. Plant Cell 20:3186–3190
Murashige T, Skoog I (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Plant Physiol 15:473–477
Ponting CP, Oliver PL, Reik W (2009) Evolution and functions of long noncoding RNAs. Cell 136:629–641
Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A (2004) Identification of mammalian microRNA host genes and transcription units. Genome Res 14:1902–1910
Rohrig H, Schmidt J, Miklashevichs E, Schell J, John M (2002) Soybean ENOD40 encodes two peptides that bind to sucrose synthase. Proc Natl Acad Sci USA 99:1915–1920
Rymarquis LA, Kastenmayer JP, Hüttenhofer AG, Green PJ (2008) Diamonds in the rough: mRNA-like non-coding RNAs. Trends Plant Sci 13:329–334
Shao F, Lu S (2013) Genome-wide identification, molecular cloning, expression profiling and posttranscriptional regulation analysis of the Argonaute gene family in Salvia miltiorrhiza, an emerging model medicinal plant. BMC Genom 14:512
Shao F, Lu S (2014) Identification, molecular cloning and expression analysis of five RNA-dependent RNA polymerase genes in Salvia miltiorrhiza. PLoS ONE 9:e95117
Song JY, Luo HM, Li CF, Sun C, Xu J, Chen SL (2013) Salvia miltiorrhiza as medicinal model plant. Yao Xue Xue Bao 48:1099–1106
Sugiyama R, Kazama Y, Miyazawa Y, Matsunaga S, Kawano S (2003) CCLS96.1, a member of a multicopy gene family, may encode a non-coding RNA preferentially transcribed in reproductive organs of Silene latifolia. DNA Res 10:213–220
Taylor CB, Green PJ (1995) Identification and characterization of genes with unstable transcripts (GUTs) in tobacco. Plant Mol Biol 28:27–38
Teramoto H, Toyama T, Takeba G, Tsuji H (1996) Noncoding RNA for CR20, a cytokinin-repressed gene of cucumber. Plant Mol Biol 32:797–808
Tran TT, Zhou F, Marshburn S, Stead M, Kushner SR, Xu Y (2009) De novo computational prediction of non-coding RNA genes in prokaryotic genomes. Bioinformatics 25:2897–2905
Voinnet O (2009) Origin, biogenesis, and activity of plant microRNAs. Cell 136:669–687
Wang M, Wu B, Chen C, Lu S (2014) Identification of mRNA-like non-coding RNAs and validation of a mighty one named MAR in Panax ginseng. J Integr Plant Biol. doi:10.1111/jipb.12239
Wen J, Parker BJ, Weiller GF (2007) In silico identification and characterization of mRNA-like noncoding transcripts in Medicago truncatula. In Silico Biol 7:485–505
Wu B, Li Y, Yan H, Ma Y, Luo H, Yuan L, Chen S, Lu S (2012a) Comprehensive transcriptome analysis reveals novel genes involved in cardiac glycoside biosynthesis and mlncRNAs associated with secondary metabolism and stress response in Digitalis purpurea. BMC Genom 13:15
Wu W, Zhu Y, Zhang L, Yang R, Zhou Y (2012b) Extraction, preliminary structural characterization, and antioxidant activities of polysaccharides from Salvia miltiorrhiza Bunge. Carbohydr Polym 87:1348–1353
Wu HJ, Wang ZM, Wang M, Wang XJ (2013) Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol 161:1875–1884
Xin M, Wang Y, Yao Y, Song N, Hu Z, Qin D, Xie C, Peng H, Ni Z, Sun Q (2011) Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol 11:61
Ye J, Fang L, Zheng HK, Zhang Y, Chen J, Zhang ZJ, Wang J, Li ST, Li RQ, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34(Web Server issue):293–297
Zhang L, Wu B, Zhao D, Li C, Shao F, Lu S (2014a) Genome-wide analysis and molecular dissection of the SPL gene family in Salvia miltiorrhiza. J Integr Plant Biol 56:38–50
Zhang Y, Yan YP, Wu YC, Hua WP, Chen C, Ge Q, Wang ZZ (2014b) Pathway engineering for phenolic acid accumulations in Salvia miltiorrhiza by combinational genetic manipulation. Metab Eng 21:71–80
Zhao G, Shi Q, Han Y, Li S, Wang C (2014) The physiological and biochemical responses of a medicinal plant (Salvia miltiorrhiza L.) to stress caused by various concentrations of NaCl. PLoS ONE 9:e89624
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415
Acknowledgments
We thank Drs. Yingjie Zhu, Caili Li, Meizhen Wang and Suzhen Chen for kindly providing technical assistance. We appreciate Prof. Xian’en Li at the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College for providing S. miltiorrhiza plants. This work was supported by grants from the Natural Science Foundation of China (Grant No. 31370327), the Beijing Natural Science Foundation (Grant Nos. 5112026 and 5152021), the Major Scientific and Technological Special Project for Significant New Drugs Creation (Grant No. 2012ZX09301002-001-031), the Research Fund for the Doctoral Program of Higher Education of China (Grant No. 20111106110033), and the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT, Grant No. IRT1150).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, D., Shao, F. & Lu, S. Identification and characterization of mRNA-like noncoding RNAs in Salvia miltiorrhiza . Planta 241, 1131–1143 (2015). https://doi.org/10.1007/s00425-015-2246-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-015-2246-z